

Subscribe to RSS
DOI: 10.1055/s-0044-1786807
Thrombophilia Screening: Not So Straightforward
Authors
Abstract
Although inherited thrombophilias are lifelong risk factors for a first thrombotic episode, progression to thrombosis is multifactorial and not all individuals with inherited thrombophilia develop thrombosis in their lifetimes. Consequently, indiscriminate screening in patients with idiopathic thrombosis is not recommended, since presence of a thrombophilia does not necessarily predict recurrence or influence management, and testing should be selective. It follows that a decision to undertake laboratory detection of thrombophilia should be aligned with a concerted effort to identify any significant abnormalities, because it will inform patient management. Deficiencies of antithrombin and protein C are rare and usually determined using phenotypic assays assessing biological activities, whereas protein S deficiency (also rare) is commonly detected with antigenic assays for the free form of protein S since available activity assays are considered to lack specificity. In each case, no single phenotypic assay is capable of detecting every deficiency, because the various mutations express different molecular characteristics, rendering thrombophilia screening repertoires employing one assay per potential deficiency, of limited effectiveness. Activated protein C resistance (APCR) is more common than discrete deficiencies of antithrombin, protein C, and protein S and also often detected initially with phenotypic assays; however, some centres perform only genetic analysis for factor V Leiden, as this is responsible for most cases of hereditary APCR, accepting that acquired APCR and rare F5 mutations conferring APCR will go undetected if only factor V Leiden is evaluated. All phenotypic assays have interferences and limitations, which must be factored into decisions about if, and when, to test, and be given consideration in the laboratory during assay performance and interpretation. This review looks in detail at performance and limitations of routine phenotypic thrombophilia assays.
The term thrombophilia is derived from the Greek words for affinity and blood clot and is used to indicate hereditary or acquired disorders of hemostasis that confer an increased tendency to form pathological intravascular thrombi.[1] [2] This is an important point, because in stark contrast to the more common forms of hemophilia A and B where bleeding is inevitable,[3] the most common thrombophilic presentation, venous thromboembolism (VTE), is a multifactorial disease and possession of a thrombophilia alone does not guarantee progression to a thrombotic phenotype.[1] [4] [5] Typical of multifactorial diseases, VTE results from single or multiple genetic, epigenetic, and/or circumstantial (i.e., acquired) predisposing factors operating in concert to precipitate the clinical presentation.[1] [5]
Acquired risk factors for VTE can be persistent or transient and provoking or nonprovoking.[1] Examples of persistent provoking risk factors are active cancer and antiphospholipid antibodies, whereas transient provoking risk factors include surgery, trauma, immobilization, severe infection, and pregnancy/puerperium.[1] [4] Older age (>60 years) and male sex can be considered as persistent nonprovoking risk factors and transient nonprovoking risk factors include oral contraception and hormone therapy.[1] [4] Genetic risk factors involve abnormalities that affect regulatory processes of coagulation, tipping the fine hemostatic balance between procoagulant and anticoagulant forces toward hypercoagulability, such as deficiencies of natural anticoagulants or gain-of-function variants of coagulation factors.[6] Hemostatic balance is mostly maintained in children and young adults who have a single genetic thrombophilia, but thrombotic disease can occur in adults as acquired risk factors become more prevalent or where multiple genetic thrombophilias coexist.[1]
The five well-established, classical inherited thrombophilias are antithrombin (AT) deficiency, protein C (PC) deficiency, protein S (PS) deficiency, activated protein C resistance (APCR) due to Factor (F) V Leiden (FVL), and the prothrombin gene mutation F2 G20210A.[6] [7] [8] Numerous genetic variants cause deficiencies of AT, PC, and PS; so routine laboratory screening for these thrombophilias generally involves a suite of phenotypic assays to demonstrate presence and severity of reduced plasma activity or concentration, rather than genetic analysis. APCR can also be demonstrated in functional phenotypic assays, although confirmation that FVL is responsible requires genetic analysis of F5.[9] F2 G20210A gives rise to elevated prothrombin levels, but functional testing for FII activity does not identify all affected patients, due to overlap between upper limits of normality and affected patients, and genetic analysis is therefore the only reliable option.[10]
Although inherited thrombophilias represent lifelong risk factors for a first episode of VTE, not all patients with an inherited thrombophilia develop VTE. As an example, only approximately 10% of heterozygous FVL carriers will develop VTE throughout their lifetime.[11] [12] Indiscriminate screening in patients with idiopathic VTE is therefore not recommended, as their presence does not necessarily predict recurrence or influence management, and testing should thus be selective,[2] [7] [13] [14] [15] [16] [17] [18] [19] [20] although even here there is not full consensus. For instance, it is generally accepted that results of inherited thrombophilia screening would not alter management for patients with unprovoked VTE,[7] [13] [17] and some contend the same holds true for most or all provoked VTE;[7] [17] however, others suggest screening has a place in patients with VTE in unusual sites and when VTE is provoked by certain nonsurgical risk factors such as pregnancy and use of combined oral contraceptives.[2] [13] Prevalence and relative risk data for the classical inherited thrombophilias are given in [Table 1].
Abbreviations: F2 G20210A, prothrombin gene mutation; FVL, factor V Leiden, VTE, venous thromboembolism.
Notes: Estimates for prevalence and relative risk vary due to different study designs and the figures given are composites from different publications.[1] [2] [17] [19] Identification of the classical thrombophilias is a relatively poor predictor of recurrent VTE, because of issues such as incomplete penetrance, concomitant acquired risk factors can be better managed or avoided, and patients are likely to be anticoagulated in response to the initial event.
Even though the debate on which clinical situations warrant screening for inherited thrombophilia remains unresolved, there is agreement that screening can be beneficial when it informs management. Another arm of the debate is that false positives and negatives with phenotypic assays contribute significantly to misdiagnoses, so screening must be undertaken in an informed manner when considered valuable to management of select patients. Indeed, the risk of misdiagnosis for AT, PC, and PS deficiencies is potentially several orders of magnitude higher than their true prevalence rate. The aim of this article is to concentrate on the details and limitations of detecting the main inherited thrombophilias with phenotypic laboratory assays to maximize diagnostic efficacy.
Antithrombin
Plasma AT activity was initially considered to have six facets, classified as ATs I to VI.[21] [22] AT I represented capacity of fibrin to bind thrombin, AT II referred to inhibition of thrombin by heparin and a cofactor, AT III was progressive heparin-independent thrombin inactivation, AT IV inhibited thrombin only when being formed from prothrombin, AT V was an abnormal thrombin inhibitor found only in pathological conditions, and AT VI was the anticoagulant effect of fibrin(ogen) degradation products. Since these activities are in fact the function of only AT III, or not AT activity at all, the name “antithrombin III” was shortened to just “antithrombin” (or AT), and the others are no longer used.[22] AT is a serine protease inhibitor, or SERPIN, that operates as a natural anticoagulant to inactivate predominantly thrombin (FIIa) and FXa, and to a lesser extent, the other active enzymes of coagulation biochemistry.[23] [24] [25] As with other SERPINS, AT functions through suicide substrate inhibition of its target serine proteases.[26] The reactive site (RS) of the AT molecule is recognized as a substrate-like sequence by the serine protease, but binding to the RS forms an irreversible 1:1 complex through formation of a covalent bond, preventing any further enzyme activity of the serine protease, which, because of the result of its own action, has “committed suicide.”
AT is an inefficient inhibitor in the absence of cofactors and is potentiated up to 1,000-fold in the presence of glycosaminoglycans in the forms of heparan sulfate and heparin.[27] AT contains a separate heparin binding site (HBS), through which heparin and heparan sulfate bind AT with high affinity via a core pentasaccharide sequence present in both molecules.[28] [29] AT exists as two isoforms, α-AT comprising approximately 90% of circulating AT, whereas approximately 10% is β-AT, the latter having higher heparin affinity due to lack of glycosylation at one of four asparagine residues (Asn135).[22] [30] Active AT is inefficient in inhibiting thrombin and FXa within the prothrombinase complex, or fibrin-bound thrombin, and inhibits mainly free thrombin.[31] The regulatory role of AT is accomplished through localization onto the vessel endothelium via surface-anchored heparan sulphate where it creates an anticoagulant milieu by scavenging potentially lethal enzymes escaping the clot that would otherwise enter the general circulation.[22] [32]
Hereditary Antithrombin Deficiency
First described in 1965 by Olav Egberg in a Norwegian family,[33] AT deficiency is an autosomal dominant disorder that significantly increases risk of VTE.[34] Although considered the strongest form of inherited thrombophilia, it presents with marked clinical heterogeneity, the risk of VTE in heterozygous individuals being increased between 5- and 50-fold.[35] Deficiencies of AT are due to alterations in the SERPINC1 gene and are divided into two types; type I is a quantitative deficiency characterized by reduced levels of functionally intact AT, and type II comprises a group of qualitative deficiencies characterized by impaired function of either the RS or HBS, or pleiotropic deficiencies affecting both.[34] [36] Homozygosity for type I and some type IIa AT deficiencies is embryonically lethal, whereas homozygosity for type II HBS defects, and mild type II RS defects, have been described.[37] Type II deficiency is more prevalent than type I in the general population, but up to 80% of symptomatic AT deficient patients have type I since it is associated with a high risk of early and recurrent VTE, and also because heterozygous type II HBS deficiencies are less thrombogenic.[36] In contrast, homozygous type II HBS deficiency is characterized by early onset arterial and venous thrombosis.[38]
Laboratory Detection of Antithrombin Deficiency
Initial testing for AT deficiency employs functional assays because reductions are apparent in all deficiencies and subtypes, whereas antigenic assays, which measure quantity irrespective of activity, cannot detect impaired function and will miss type II defects if performed in isolation.[39] Type I deficiencies typically have concordantly reduced activity and antigen levels. Antigen levels are often normal in type II RS and HBS deficiencies, the antigen level being approximately twice that of the activity. Type II pleiotropic deficiencies tend to exhibit moderate reductions in activity and antigen values, yet the activity is still usually lower than the antigen.[39]
Antithrombin Activity Assays
Routinely used AT activity assays are amidolytic methods with chromogenic end points, commonly performed on automated coagulation analyzers with commercially produced reagent kits.[34] [39] Excess heparin and a target protease, either thrombin or FXa, are added to citrated plasma to ensure that every AT molecule is occupied by a protease, which is inhibited. The heparin potentiates the reaction to reduce assay time and facilitates detection of heparin-binding defects. Since the protease is used in excess, it is residual protease that is reacted with a chromogenic peptide substrate that mimics its natural substrate to generate a colored product, para-nitroaniline, the intensity of which is inversely proportional to the AT level.[34] [39] [40] An assay employing thrombin inhibition is depicted in [Fig. 1].

Since the end point of the assay is color generation, plasma samples that are hemolyzed, icteric, or lipemic (HIL) can interfere with end point measurement. In practice, this is rarely a problem because plasmas are typically diluted 1:20 prior to analysis, and where innate plasma coloration remains problematic, it can be overcome using sample-specific blanks. One difficulty with assays based on thrombin inhibition is that these are also inhibited by heparin cofactor II (HCII), leading to overestimation of AT levels.[40] This can be overcome by replacing human thrombin with bovine thrombin, where HCII interference is significantly minimized, or use of FXa as target protease where HCII interference is nonexistent.[40] Prolonged incubation time with heparin and protease can reduce sensitivity to type II HBS defects, particularly when using bovine thrombin-based assays, and the incubation period should be no more than 30s, unless a given reagent has been validated to detect type II HBS at a longer incubation.[39] [40] Kovács et al reported FXa-based assays to be more sensitive to HBS defects than thrombin-based assays.[41] Chromogenic AT activity assays are unaffected by anticoagulation with vitamin K antagonists (VKA) or heparins, fondaparinux has minimal and clinically insignificant effects on FXa-based assays, but direct thrombin inhibitors (dabigatran, argatroban, bivalirudin) and direct FXa inhibitors (DFXaI) (rivaroxaban, apixaban, edoxaban) can falsely elevate results in FIIa- and FXa-based assays, respectively.[39] [42] [43] Thus, a dose-dependent factitious increase in measured AT levels occurs in the presence of direct oral anticoagulants (DOAC), which can occur even at trough drug levels.[44] This can be overcome by interrupting DOAC therapy for 2 to 3 days prior to sample collection, which in reality is rarely clinically feasible, or by pretreating DOAC-containing plasmas with DOAC-neutralizing adsorbents composed of activated charcoal.[42] [45]
Antithrombin Antigen Assays
Antigen assays are unaffected by anticoagulation and would serve to detect or exclude type I deficiency, the most common subtype in symptomatic patients, but not the qualitative type II deficiencies.
A reduced activity result is sufficient to confirm the presence of a deficiency, and distinguishing between types I and II through concordance or discordance with an antigen result, respectively, does not necessarily affect management, except that some type II defects have lower thrombotic risk,[22] so some laboratories choose to routinely subtype.[40]
Radial immunodiffusion, Laurell “rocket” immunoelectrophoresis, and enzyme-linked immunosorbent assays (ELISA) have all been used to quantify AT antigen.[22] [39] [40] Such assays can be technically demanding, time consuming, and have poorer precision than automated activity assays. Recent years have seen the availability of rapid automated latex immunoassays (LIA) for AT antigen, with low imprecision.[39] [46] Briefly, microscopic latex particles coated with antibodies to AT react with AT in the test sample, leading to agglutination of the latex particles. The degree of agglutination is directly proportional to the AT antigen level and is measured by light absorption, typically at 620 mn. AT antigen assayed with LIA can be falsely reduced in pregnancy, or falsely elevated by dysproteinemia or high levels of rheumatoid factor, which can be circumvented with higher sample dilutions.[39] [47] AT antigen assays should be quantified in the same units as activity assays (IU/mL or IU/dL) to be able to directly compare values when assessing for concordance or discordance.
Distinguishing between Type II Subtypes
If a type II subtype is suggested by activity/antigen discordance, assays are available to demonstrate whether the HBS or RS are dysfunctional. The progressive chromogenic AT activity assay assesses inhibition of thrombin or FXa independently of heparin to specifically isolate RS function, using the same chromogenic substrates as in standard assays to detect residual enzyme.[39] [40] [48] The absence of heparin in the assay necessitates incubation times longer than 30s, which can be between 3 minutes and an hour. By not selectively potentiating AT with heparin, the assay has reduced specificity arising from the action of other inhibitors such as α-1-antitrypsin and α-2-macroglobulin.[39] [40] Plasmas containing HBS defects generate normal results with the progressive assay but reduced results with standard assays, whereas results of both assays are reduced with RS defects and type I deficiencies.
Isolating HBS function can be done with two dimensional crossed immunoelectrophoresis where plasma is initially electrophoresed into a heparin-containing agarose gel. The gel is then rotated by 90 degrees and the AT is electrophoresed into a second agarose gel containing a precipitating antibody to AT, the AT mobility being visualized by drying and staining the gel.[40] [49] AT bound to heparin has increased electrophoretic mobility and migrates further in the first dimension than AT with an HBS defect, thereby forming two distinct peaks in the second dimension ([Fig. 2]). This is a time-consuming and technically demanding procedure and consequently available only in a few specialist centres. A more straightforward alternative is the heparin-antithrombin binding (HAB) ratio where plasma is assayed with a standard activity assay at short (30s) and prolonged (5 minutes) incubations and a ratio derived from the two values.[50] Using the result from the prolonged incubation time as denominator, the reduced sensitivity to HBS defects at longer incubations results in higher values than at short incubations, leading to low ratios with HBS defects. A limitation of this assay is that HBS defects devoid of heparin binding ability, as opposed to reduced capacity which can be exaggerated with prolonged incubation, cannot respond to the HAB ratio principle.

Unusual Antithrombins
At first sight, detecting AT deficiencies through careful performance of routinely available activity assays might appear straightforward. However, thrombin-based and FXa-based assays are not entirely interchangeable and misdiagnosis can occur with some type II deficiencies. AT Cambridge II (p.Ala416Ser) and AT Denver (p.Ser426Leu) are RS defects that commonly return normal values in FXa-based assays but mild to moderately reduced results in thrombin-based assays.[40] [51] [52] [53] This is because AT Cambridge II reacts with both thrombin and FXa, but only FXa becomes trapped and inhibited, and AT Denver has a significantly more reduced inhibition rate constant for thrombin than FXa in the presence of heparin. Because the RS of AT Cambridge II inhibits FXa, the progressive activity assay is normal, which in the presence of a reduced result with a standard thrombin-based assay, can erroneously suggest an HBS defect. A short incubation time in thrombin-based assays improves sensitivity to AT Cambridge II.[40] Furthermore, AT Cambridge II can return results within the lower part of the reference range for thrombin-based assays, and phenotypic detection relies on additionally performing an antigen assay (on the same sample) to evidence a reduced activity/antigen ratio.[40] Some centres routinely perform antigen assays on low-normal activity levels.[40] AT Stockholm (p.Gly424Asp) is another RS defect that manifests in human and bovine thrombin-based assays, but FXa-based assays can sometimes overestimate the level and generate a normal result.[54]
AT Glasgow (p.Arg425His) is an RS defect with reduced thrombin and FXa inhibitory activity despite also possessing increased heparin affinity.[40] [55] Reduced levels are obtained in FXa-based assays but prolonged incubation time (>1–2 minutes) with thrombin-based assays can normalize the result, even if bovine thrombin is employed.[39] [40] AT Wobble (p.Thr117Lys) is a conformationally unstable variant that will give reduced levels in FXa-based and bovine thrombin-based assay but can give normal or reduced levels in human thrombin-based assays.[40] [56]
Description of transient AT deficiencies has led to the suggestion that the prevalence of AT deficiency may be underestimated.[57] This is partly due to variable success rates in detection of the type II deficiencies described above, and also because variants exist that are susceptible to environmental factors that precipitate transition to altered molecular forms lacking inhibitory activity.[57] AT Rouen VI (p.Asn219Asp) was first reported to undergo spontaneous polymerization and formation of monomeric latent AT, occurring slowly at 37°C but markedly accelerated at 41°C, explaining the association of thrombotic episodes with pregnancy-associated pyrexial infections.[58] Subsequently reported symptomatic patients were not pyrexial but almost all were pregnant.[59] AT Rouen VI can present with normal or reduced AT levels or even normal antigen but reduced activity. Other ATs conformationally unstable under stress that may give reduced, borderline, or normal phenotypic results include AT Wibble (p.Thr117Met), AT Truro (p.Glu269Lys), and AT Budapest 5 (p.Pro439Thr), which are prone to latent transformation, and AT Dublin (p.Val30Glu), and the p.Thr250Ile and p.Ile386Thr variants, which are prone to polymerization.[56] [57] [60] [61] AT Wobble (p.Thr117Lys) is a significantly more unstable variant that undergoes spontaneous transition to the latent form, presenting as an apparent type I deficiency with moderately reduced activity and antigen levels due to cumulative loss rather than a deficiency of reduced synthesis.[56] The index patient presented with an ileofemoral thrombosis at 10 years of age in association with a preceding pyrexial respiratory infection thought to have induced sudden, rapid confirmational change. Two new AT variants have recently been described, p.Glu227Lys and p. Asn224His, in four unrelated patients with early and recurrent thrombosis, whose thrombin- and FXa-based activity assays, and antigen assays are almost always normal, but they have impaired anti-FVIIa activity, for which standardized commercial assays are not available.[62] The inconsistent ability of routine AT assays to identify these types of AT deficiency emphasizes that genetic analysis is sometimes the only reliable route to detect some AT deficiencies.
Acquired Causes of Antithrombin Deficiency
Before abnormal AT phenotypic results are translated into a diagnosis of a hereditary deficiency, the possibility of an acquired deficiency must be considered. Acquired AT deficiency due to impaired synthesis can occur in liver disease, poor nutrition, L-asparaginase therapy, inflammatory bowel disease, and severe burns.[22] [39] Consumptive acquired AT deficiency can be seen in acute thrombosis, surgery, disseminated intravascular coagulation (DIC), heparin therapy (due to increased clearance), widespread malignancy, preeclampsia, hemolytic transfusion reaction, extracorporeal membrane oxygenation, and acute myeloid leukemia.[22] [39] Increased loss of AT may be encountered in nephrotic syndrome, chylothorax, and the dilutional effects of hemodialysis, plasmapheresis, and cardiopulmonary bypass.[22] [39] Although not strictly a deficiency, it should be noted that AT levels are lower than adults at birth and up to 6 months of age, although physiologically this is typically balanced by lower levels of procoagulant factors in this age group.[39]
Protein C
PC is the vitamin K-dependent zymogen of the serine protease activated protein C (APC), which serves a critical role in regulation of thrombin generation by inactivating FVa and FVIIIa, the cofactors for the prothrombinase and tenase complexes, respectively.[63] PC is named as such because elution of vitamin K-dependent factors from a DEAE–Sephadex column generated four peaks, which when labelled A to D, PC comprised the third peak.[64] The PC pathway is a multicomponent process involving proteins that activate, modulate, and inhibit APC.[65] PC is localized to the vessel endothelium via endothelial protein C receptor (EPCR), which presents PC to the thrombin:thrombomodulin (TM) complex for activation.[66] Thrombin bound to endothelium-anchored TM undergoes conformational alteration such that its procoagulant properties are abolished and it gains the ability to activate PC and thrombin-activatable fibrinolysis inhibitor.[67] Thrombomodulin functions as a cofactor to thrombin, accelerating the rate of PC activation >1,000-fold. Activation is further enhanced by EPCR, chondroitin sulfate, and platelet factor 4, resulting in accumulation of APC at the site of injury where activated platelets are promoting coagulation. The APC is released by EPCR whereby, unlike AT, it enters the forming clot by binding to the phospholipid surface of activated platelets.[68] APC combines with its vitamin K-dependent, nonenzymatic cofactor, PS, on the phospholipid surface to inactivate FVa by limited proteolysis, whereas FVIIIa inactivation requires APC-cleaved FV to operate as an APC cofactor in synergy with PS.[69] [70] This pathway reveals FII and FV adopting both procoagulant and anticoagulant functions, classifying them as Janus-faced proteins.[69] [71] Janus was the ancient Roman god of transitions, beginnings and endings, and entrances and exits, who is depicted in art with two faces looking in opposite directions, one facing the past and the other the future, representing different perspectives.[72] The month of January and one of Saturn's moons are named after him. APC is inactivated by the nonspecific inhibitors PC inhibitor, α-1-antitrypsin, and α-2-macroglobulin, resulting in a half-life in the circulation of about 20 minutes.[65] [73]
Hereditary Protein C Deficiency
PC deficiency arises through alterations in the PROC gene and is predominantly an autosomal dominant disorder, with variable penetrance.[68] [70] Similar to AT deficiency, PC deficiency occurs either as a type I quantitative disorder presenting with reduced amounts of normally functional PC, or type II qualitative disorders with usually normal levels of dysfunctional PC.[70] Type I deficiency is the most common, comprising up to 80% of all cases.[68] [74] Most type II deficiencies involve defects in the activation site or the active site, termed type IIa, whereas type IIb deficiencies involve reduced binding to substrates, cofactors, or phospholipid and comprise no more than one percent of all PC deficiencies.[74] Simple heterozygous PC deficiency with mild to moderately reduced PC levels can range clinically from asymptomatic, to typical VTE in adulthood, to a severe thrombophilia with recurrent thromboses.[63] Conversely, the autosomal recessive severe congenital protein C deficiency (SCPCD) typically presents with potentially fatal purpura fulminans and DIC within 72 hours of birth and undetectable plasma PC.[75] Rarely, less severe SCPCD presents with delayed onset at about 10 months of age, or individuals surviving into adulthood before experiencing VTE.[75] [76] Observed prevalence of SCPCD is lower than predicted incidence, suggesting fetal demise or undiagnosed perinatal deaths.[75] A rare complication of treating patients with heterozygous PC deficiency with VKA anticoagulation is so-called warfarin-induced skin necrosis.[68] [77] PC has a shorter half-life than other vitamin K-dependent factors, causing an exaggeration of the deficiency in the first 24 to 48 hours of VKA therapy, resulting in a relatively hypercoagulable state. In some cases, this leads to excessive clotting in the dermal microvasculature, resulting in skin necrosis.
Laboratory Detection of Protein C Deficiency
Initial testing for PC deficiency also employs functional assays as levels will be low in all subtypes. Antigen assays can be employed to distinguish between types I and II, although it is not essential as clinical expression does not differ and it does not affect management.[70]
Protein C Activity Assays
Two types of assays are routinely available for quantifying PC activity, based on either clotting time or chromogenic end points ([Fig. 3]).[78] [79] Both assay types have a common starting point, which is activation of PC in citrated plasma by an exogenous fast acting direct PC activator isolated from venom of the Southern Copperhead snake (Agkistrodon contortrix contortrix), a pit viper from southern United States.[80] The venom activator, known as Protac, is used in preference to the physiological activator of PC, thrombin, as thrombin has procoagulant activity additional to the test system itself, PC activation is incomplete without TM, and thrombin reacts with chromogenic substrates.[80] Some early PC activity assays did employ thrombin in complex with TM, but they were complex and expensive.[81] Rapid activation of PC by Protac minimizes interference by inhibitors of APC.[81]
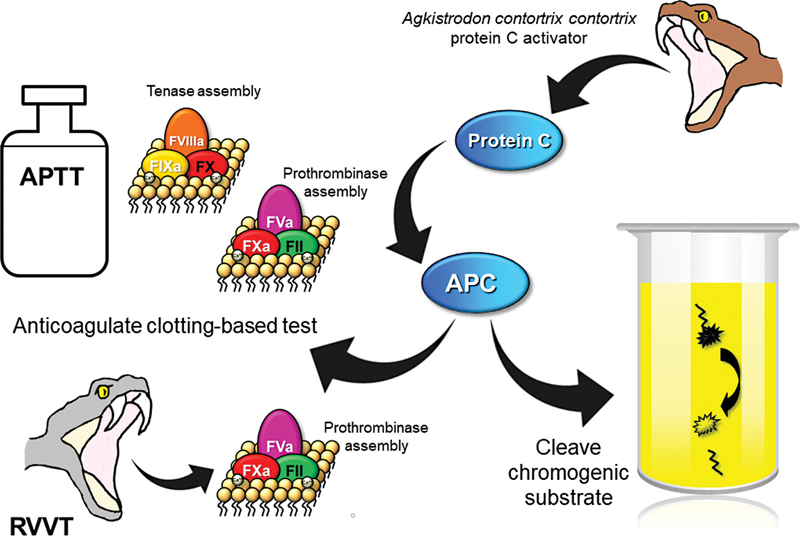
Clotting-based PC activity assays employ either activated partial thromboplastin time (aPTT) or Russell's viper venom time (RVVT) to achieve the fibrin clot end point.[78] [79] [80] [81] Activation of PC by the Protac initiates an anticoagulant effect, prolongation of the clotting time being proportional to PC activity. In the intrinsic pathway-based aPTT assays, the APC inactivates both FVIIIa and FVa, whereas the direct FX activation in RVVT permits downstream inactivation of only FVa. As with any clotting-based assay used to isolate a specific abnormality, the design necessarily assumes all else about the patient's coagulation is normal. Therefore, other causes of elevated clotting times, such as factor deficiencies and therapeutic anticoagulation, including DOACs, can cause overestimation of PC activity.[42] [79] [81] Lupus anticoagulants (LA) can have variable effects in that overestimation of PC activity can occur through interference with phospholipid-dependent coagulation reactions, adding to the anticoagulation by APC, but underestimation can alternatively arise if the LA directly interferes with the anticoagulant activity of APC.[79] [81] [82] Phospholipid composition of the reagents dictates how LA affects each assay. Elevated PS can lead to overestimation of PC activity in clotting-based assays.[74] APCR can give rise to falsely reduced PC activity in clotting-based assays, particularly at low dilutions, and elevated FVIII can lead to PC underestimation in aPTT-based assays. Interferences can be reduced or eliminated by diluting test plasma in PC-deficient plasma, although this approach affects the lower limit of quantification, and some reagents additionally contain heparin neutralizers. DOAC neutralizing adsorbents can be used to negate DOAC interference in PC clotting-based assays.[83] If specificity may be hampered by the presence of interfering factors, the assay can be performed using multiple sample dilutions to increasingly reduce interference, as is done for coagulation factor assays.[79] [84]
Chromogenic PC activity assays assess PC function through cleavage of a chromogenic substrate by the venom-activated APC, the intensity of the colored product being directly proportional to PC activity.[78] [81] One limitation of these assays is that the chromogenic substrates can also be cleaved by enzymes other than APC, such as thrombin, FXa, kallikrein, FXIIa, and plasmin, which may be present in in vitro activated or clotted blood samples arising from difficult venepuncture, or may be present in vivo in DIC or therapeutic fibrinolysis, thereby leading to false elevation of PC activity.[79] [81] Cold-activated plasma can cause overestimation of PC in chromogenic assays as it contains kallikrein and FXIIa.[85] The finding of an exceptionally elevated value (i.e., >200 IU/dL) should prompt suspicion of activation, which can be confirmed by running the assay with water in place of Protac to detect non-APC substrate cleavage.[79] Underestimation of PC can occur in samples with significant HIL.[74] PC levels are inevitably reduced in VKA therapy, although measured activity level can differ between clotting-based and chromogenic assays, levels tending to be higher with the latter because it measures a vitamin K-induced carboxy PC molecule that is nonfunctional in vivo.[74] [86] Furthermore, the chromogenic assay does not measure all aspects of PC function, which as well as affecting relative sensitivity to the overall effects of VKA anticoagulation, feeds into a separate issue about assay choice. Chromogenic assays are recommended as first-line assays, because they are not only unaffected by the coagulation aberrations that interfere with clotting-based assays, conferring greater specificity, but also, the long clotting times encountered in clotting assays impart a higher degree of imprecision than seen with chromogenic assays.[68] [79] However, the principle of the chromogenic assay limits its ability to detect type II defects because interaction of APC with cofactors (PS and FV) and substrates (FVa and FVIIIa) through Ca2+-dependent binding to phospholipid to achieve anticoagulation does not occur in chromogenic assays. Consequently, chromogenic assays cannot detect type IIb defects, which are due to mutations affecting the PC Gla domain.[79] [87] [88] [89] Clotting-based assays can detect all PC deficiency types, although variable sensitivity to the Asn2Ile substitution, causing a type IIb deficiency, has been described.[90] The rarity of type IIb defects helps to justify use of chromogenic assays as first-line tests, although the preponderance of chromogenic assays in routine diagnostic use could explain why these deficiencies are not encountered as often as they might be. Seidel et al reported that 20/102 patients with PROC variants had normal results with chromogenic testing but reduced PC activity with clot-based testing, suggesting that additional testing with clot-based assays should be employed in clinically appropriate patients.[89] Given that thrombophilia testing is becoming more selective, accuracy of screening is important for those who require it, which could be improved with adoption of clotting-based assays in addition to chromogenic assays for PC deficiency detection. This can be either locally as joint first-line assays, as second-line assays, or involve use of a referral laboratory for appropriate patients.
Protein C Antigen Assays
Although it is rarely necessary to subtype PC deficiency for management purposes, it can be useful in family studies.[79] [81] Radial immunodiffusion, Laurell “rocket” immunoelectrophoresis, and enzyme-linked-fluorescent assays have all been used to quantify PC antigen, ELISA being the most commonly employed, and LIAs are available in some countries.[79] [91] Subtyping of PC deficiency based on laboratory data is shown in [Table 2].
↓: decreased.
Acquired Alterations in Protein C Levels
Acquired PC deficiency is more common than congenital deficiencies and can be due to impaired synthesis in liver disease, L-asparaginase therapy, vitamin K deficiency, and VKA anticoagulation.[68] [81] Attempting phenotypic assessment for hereditary PC deficiency during VKA therapy is futile, and calculating a ratio of PC level versus FVII level (as it has a similar half-life) is unreliable, not least because PC clotting, chromogenic, and antigen assays give different results on VKAs.[68] [78] Acquired PC deficiency arising from increased turnover/consumption occurs in acute thrombosis, DIC, renal insufficiency, postoperative state, adult respiratory arrest syndrome, sepsis, autoimmune antibodies to PC (with or without antiphospholipid syndrome [APS]), plasma exchange, malignancy, and massive hemorrhage.[68] [78] [92] Elevated PC levels can occur in pregnancy or postpartum, postmenopause, hormone replacement therapy (HRT), oral contraception, nephrotic syndrome (can also be decreased), diabetes, and in association with elevated lipids.[68] [78] [93] [94] [95]
Interpreting Results against a Reference Range
A typical reference range for phenotypic PC assays would be 75 to 130 IU/dL, although ranges are reagent, analyzer, and population specific and should be locally derived.[79] [96] [97] [98] Crucially, PC levels are significantly lower than adults in infancy and young childhood and may not normalize to adult levels until 16 years of age.[96] [97] [98] [99] [100] [101] [102] Studies on pediatric hemostasis parameters commonly generate ranges for age groups, such as Day 1, Day 3, 1 to 6 months, 6 to 12 months, 1 to 5 years, 6 to 10 years, 11 to 16/18 years, which demonstrate gradual increases in PC with age,[97] but between-platform variability prevents recommendation of generic ranges. It is logistically almost impossible for every laboratory to generate its own ranges for every hemostasis parameter for each age group and the best option is to adopt published ranges employing at least the same reagents, which would commonly be used with a manufacturer-matched analyzer.[79] [97]
Some studies have reported a modest rise in PC levels during adulthood,[103] [104] [105] a more significant rise being seen in older women from the effects of menopause and HRT, whereas others suggest these elevations are less marked, or not even present.[79] [106] [107] Even if present, the age-related increases, and other acquired increases described above, do not necessarily mean the PC level will exceed the upper limit of the reference range, but they have potential to raise the level of a mildly deficient patient above the lower limit of normal, where a deficiency would be missed. Additionally, Allaart et al reported that up to 15% of genetically proven PC-deficient heterozygotes can not only return PC levels within the lower part of a reference range,[108] which may in part be due to assay choice,[89] but also reflects that any assay cutoff represents some degree of compromise between sensitivity and specificity.
Protein S
PS, so named as it was isolated and characterized in Seattle, Washington,[109] is the vitamin K-dependent, nonenzymatic cofactor for both APC and tissue factor pathway inhibitor (TFPI), thereby contributing to downregulation of coagulation in both the initiation and propagation phases.[110] [111] [112] Similar to the way APC-cleaved FV operates synergistically with PS in cofactor activity for FVIIIa inactivation by APC, the splice isoform of FV, FV short, enhances the TFPIα-cofactor activity of PS with respect to FXa inhibition.[113] [114] Approximately 60% of plasma PS circulates noncovalently bound to an inhibitor of the complement system, C4b-binding protein (C4BP), in a 1:1 molar ratio, with any molar surplus (∼40%) circulating as free PS (FPS).[114] [115] In conjunction with FPS, APC inactivates FVa through cleavage of the FVa heavy chain at Arg306 and Arg506, and while bound PS is considered largely inert with respect to anticoagulant function, this may because its ability to stimulate APC-catalyzed cleavage at Arg306 is counteracted by PS-C4BP inhibition of APC-catalyzed cleavage at Arg506.[111] [116] Bound PS contributes to regulation of the complement system by localizing C4BP on negatively charged phospholipid membranes of apoptotic cells.[5]
Hereditary Protein S Deficiency
Hereditary deficiencies of PS arise from alterations in the PROS1 gene, which are predominantly autosomal dominant disorders.[117] Quantitative PS deficiencies are the most common, of which there are two types.[118] Type I deficiencies are characterized by reductions in both total PS (TPS) (bound + free) and FPS, whereas type III deficiencies exhibit normal TPS but reduced FPS. The quantitative reduction of FPS in both cases is consequently accompanied by reduced PS activity. Type I and type III deficiencies often occur within the same family associated with the same mutation, suggesting that they are phenotypic variants of the same genetic disorder, the differences being accounted for by C4BP levels.[118] [119] Type II PS deficiency is a qualitative disorder characterized by normal levels of TPS and FPS and reduced PS activity.[120] Type I deficiency accounts for up to 80% of PS deficiencies, type III for up to 20%, and type II up to 5%.[120] Not all individuals with PS deficiency experience VTE, the risk depending on multiple gene–gene or gene–environment interactions, and those with more marked PS reductions have a higher risk.[117] [121] Homozygous or compound heterozygous PS deficiency is extremely rare and potentially fatal, typically presenting with massive VTE or neonatal purpura fulminans. Warfarin-induced skin necrosis has been reported in some PS-deficient patients.[122] [123] [124]
Laboratory Detection of Protein S Deficiency
The recommended assay for initial assessment for PS deficiency is an antigenic determination of FPS.[118] This is counterintuitive because such assays cannot detect type II PS deficiencies, yet because they are considered to be rare, and currently available PS activity assays are burdened with numerous interferences and only moderate specificity, FPS antigen assays are considered a suitable starting point.[74] [118] [120] Again, the question of actual frequency of a rare subtype arises when we don't test for it as often as for the others. Assays for TPS will not detect type II or III deficiencies, and they contribute little to clinical management since there is no difference in clinical phenotype between the subtypes.[74] [118] [120] However, they are useful in family studies and can alert to the possibility of some acquired PS deficiencies where TPS is normal and FPS is reduced.[74] [118]
Protein S Activity Assays
Biological and analytical variables make measurement of PS activity to detect hereditary defects a difficult proposition.[120] Currently available commercial PS activity assays are clotting-based and employ measurement of APC-cofactor activity but not TFPIα-cofactor activity of PS. They are based on prothrombin time, aPTT, RVVT, or addition of exogenous FIXa or FXa, for in vitro clot formation, and APC is either exogenous or generated by Protac ([Fig. 4]). Use of Protac in a PS activity assay relies on the test plasma containing normal levels of PC, which may not be the case in the patient population being tested, and variable levels of endogenous PC reduces standardization, although it can be overcome by predilution in PS-deficient plasma. A chromogenic assay specifically designed to detect type II deficiencies has been described but is not commercially available.[125] Inevitably, aPTT/FIXa based assays will measure APC-cofactor activity in the inactivation of both FVIIIa and FVa, whereas the other assays will only inactivate FVa. In the clotting-based assays, the degree of PS-dependent anticoagulation prolongs the clotting time in direct proportion to the PS activity.

Elevated FVIII can shorten clotting times in aPTT/FIXa-based assays, artifactually decreasing PS activity results, which can also occur in all the assays in the presence of elevated prothrombin.[117] [120] [126] Underestimation can also occur where APCR is present (i.e., FVL), and factor deficiencies can cause artifactual overestimation. These interferences have contributed to the acceptance of only moderate specificity with PS activity assays, and while commercial assays commonly employ dilution of test plasma in PS-deficient plasma to reduce or eliminate the effects of these variables,[120] interference can still occur in some samples.[127] Inclusion of exogenous FVa in the reagents has been used to negate the effects of APCR.[126] As with clotting-based PC activity assays, LA can cause over- or underestimation of PS activity levels depending on the nature of interference in a given assay by a given antibody.[74] [128] [129] Dilution in PS-deficient plasma may eliminate the effects of some LA, but more potent or higher titer antibodies may overcome the dilution, so knowledge of presence/absence of LA in a sample being tested for PS activity, and apparent potency, are valuable when interpreting PS activity results. This is further complicated by the existence of genuine acquired FPS deficiency in some patients with antiphospholipid antibodies,[128] [130] [131] [132] although such acquired deficiencies can be expected to be reflected in a concordantly reduced FPS antigen value. Fortunately, PS activity assays tend to be performed alongside other assays that will confirm/refute the presence of potential interferences such as APCR and LA, and significant factor deficiencies (which are unlikely in the population being tested) will manifest in coagulation screening, emphasizing that PS activity results should not be interpreted in isolation. External quality assurance schemes have reported between-assay variability for PS activity assays in detection of PS deficiency,[133] [134] which may be at least partly due to use of manufacturer's reference ranges instead of locally derived reference ranges.[135] Falsely decreased values have been reported with multiple assays in normal individuals that are then normal when repeated on a new sample,[13] [136] and conversely, false-normal activity levels have been reported in samples with reduced FPS antigen.[137] These phenomena are considered to be unexplained, but biological variation and assay interferences are likely explanations for at least some of the apparent discrepancies.
PS is of course reduced on VKA anticoagulation, so testing for hereditary PS deficiency with any phenotypic assay in that situation is also futile, and commercial assays tend to include reagent-integral heparin neutralizers capable of quenching heparins at therapeutic levels. DOACs interfere with PS activity assays, resulting in overestimation, and DOAC adsorbents are a potential solution.[127] [138]
Free Protein S Antigen Assays
Laurell “rocket” immunoelectrophoresis and then ELISA were the mainstay assays for FPS,[126] but they have largely been superseded by LIAs as they are rapid, more precise, and easily automated.[120] Prior to the availability of monoclonal antibodies that are specific for FPS because they are directed to the C4BP binding site, PS bound to C4BP had to be precipitated out from plasma by addition of 3.75% polyethylene glycol (PEG), leaving only FPS in the supernatant to be detected in the ELISA or immunoelectrophoresis assay with antibodies that recognize both bound PS and FPS.[126] Assaying untreated plasma in the same run additionally generated a TPS result, and such assays remain viable alternatives for laboratories without access to full automation, although reproducibility when using PEG precipitation can be poor.[139] [140] Delay in addition of diluted test plasma to the ELISA microplate, prolonged incubation periods, and incubation temperatures that are too high, have been reported to lead to overestimation of FPS in PS-deficient patients.[141] ELISAs specifically for FPS estimation employ either monoclonal antibodies or C4BP to capture FPS, whereupon it is reacted with suitably labelled antibodies to facilitate end point measurement.
The LIAs used to quantify FPS antigen employ the same principle as those described above for AT, using latex particles coated with two monoclonal antibodies to capture FPS and cause agglutination, or latex particles coated with purified C4BP to capture only FPS, followed by addition of a second reagent containing latex particles coated with a monoclonal antibody to human PS to initiate agglutination in direct proportion to the FPS concentration. High levels of rheumatoid factor, heterophilic antibodies, and HIL, and significant platelet contamination of plasma, are potential interferences leading to overestimation. The LIAs are not plagued by the numerous hemostatic variables that affect PS activity assays, rendering them more accurate in detecting true deficiencies.[120] Subtyping of PS deficiency based on laboratory data is shown in [Table 3].
Abbreviation: APC, activated protein C.
↓: decreased.
Given that type II PS deficiencies are considered to be rare, FPS antigen assays are often referred to as surrogate markers of PS function. While it is true that reduced FPS should, by definition, be accompanied by concordantly reduced functional levels, it must be emphasized that FPS antigen assays cannot detect the reduced function of type II PS deficiencies. If type II deficiencies truly comprise no more than 5% of all PS deficiencies, then use of FPS antigen alone does indeed detect the majority of PS deficiencies.[74] [120] A choice to exclude PS activity testing from a thrombophilia screening repertoire is a choice to miss type II deficiencies, which must be particularly borne in mind in specific populations where type II deficiency is more prevalent.[118] A study by Duebgen et al on 49 patients with PROS1 mutations revealed four (8.1%) as having reduced PS activity but normal FPS antigen, from which the authors concluded that functional assessment is necessary in addition to FPS antigen measurement in phenotypic detection of PS deficiency.[142] Type II deficiencies with only defective TFPIα cofactor activity, or pleiotropic deficiencies, have yet to be described.[143] Their identification would warrant development of TFPIα cofactor activity PS assays suitable for routine analysis, and an adjustment to the current classification has been proposed.[143]
Total Protein S Antigen Assays
Few laboratories routinely perform assays for TPS as they add cost and time/resource demands while contributing little or nothing to clinical management, although some centres reflex to TPS testing if FPS antigen and/or PS activity are reduced. Commercial and laboratory-developed assays based on ELISA or LIA have been developed for quantifying TPS antigen.[74]
Unusual Protein S Variants
PS Heerlen (p.Ser501Pro) is a frequently encountered missense mutation in the Caucasian population (∼0.5%) causing a type III deficiency.[118] Originally considered to be a neutral variant with little or no pathogenicity,[144] subsequent reports have suggested that it may be significant when associated with other acquired or genetic VTE risk factors[145] [146] [147] or have demonstrated an association with VTE risk.[148] Heterozygotes exhibit mildly reduced FPS antigen levels,[145] [148] but they may also be normal,[145] [148] whereas homozygotes have lower FPS levels and may have reduced TPS, thereby presenting as a type I deficiency.[148] [149] PS Heerlen has a reduced half-life in vivo, which has been linked to the mild and marked FPS reductions in heterozygotes and homozygotes, respectively.[150] [151] Heeb et al reported that the lack of clear evidence (at the time) for increased VTE risk could be due to increased direct anticoagulant activity of PS Heerlen in complex with C4BP,[152] whereas Suchon et al hypothesized that since the mutation is located in the TFPI binding domain, impaired TFPI function could contribute to thrombosis risk with PS Heerlen.[148]
PS Tokushima (p.Lys196Glu) is prevalent in the Japanese population, causing a type II deficiency with an increased risk for DVT of 4- to 8-fold in heterozygotes.[118] [125] [153] Being a type II deficiency it can only be detected phenotypically with activity assays, yet standard PS activity assays, of different principles, do not always detect PS Tokushima.[91] [118] Interestingly, the noncommercially available chromogenic assay has been shown to be sensitive to PS Tokushima.[125]
Acquired Causes of Protein S Deficiency
As with PC, acquired PS deficiency due to impaired synthesis can occur in liver disease, L-asparaginase therapy, vitamin K deficiency, and VKA anticoagulation.[154] Increased turnover/consumption leading to PS deficiency can occur in acute thrombosis, DIC, recent surgery, and nephrotic syndrome.[118] [154] There is urinary loss of only FPS in nephrotic syndrome, which in conjunction with elevated C4BP levels, leads to an acquired type III PS deficiency.[155] Similarly, elevated C4BP levels have been linked to acquired type III deficiency in diabetes mellitus,[156] whereas nonenzymatic glycosylation leading to reduced PS function, and also reduced AT and PC function, has been suggested to cause deficiencies in poorly controlled diabetes mellitus.[157] Chronic inflammation, such as in systemic lupus erythematosus (SLE), and some acute phase reactions, can cause sufficiently elevated C4BP levels to rob the plasma of FPS and appear as an acquired type III deficiency.[118] This phenomenon is not ubiquitous because synthesis of the α-chain of C4BP increases more than that of the β-chain in inflammatory disorders, and the binding site for PS is located on the C4BP β-chain.[115] Acquired type III deficiency can also be seen in oral contraception, estrogen replacement therapy, and pregnancy.[118] FPS and PS activity can fall significantly during pregnancy, where even TPS can be reduced at later stages.[158] [159] Separate reference ranges have been reported for different stages of pregnancy, yet they are platform-specific and testing for PS deficiency should be postponed until at least 6 weeks' postpartum.[158] [159] [160] [161] Autoimmune PS deficiency has been reported in infections, particularly HIV[162] [163] and chickenpox,[164] [165] and in association with multiple myeloma,[166] Behçet's disease,[167] antiphospholipid antibodies,[168] [169] [170] and SLE,[170] [171] [172] commonly leading to reduced FPS and/or activity but normal TPS. Numerous hemostatic changes have been reported in sickle cell disease such that it can be considered an ongoing hypercoagulable state, or even an inherited thrombophilia,[173] which can feature acquired moderate reductions of PC and PS during the “steady state,” and further reductions during acute pain episodes.[174] There are conflicting reports on AT levels in sickle cell disease.[174]
Protein S Reference Ranges
Plasma TPS and FPS levels are lower in women than in men, and in premenopausal than in postmenopausal women.[105] [106] [126] [175] Consequently, some laboratories adopt adult sex-specific reference ranges for PS assays. However, some reports suggest the differences are insufficient to warrant separate ranges and the decision to implement sex-specific ranges should be made locally,[74] [118] ideally based on population- and assay-specific data.
Similar to PC, PS levels have been reported to be lower during childhood,[96] [99] [100] and particularly during the first 3 to 12 months of life.[97] [101] Actual ranges for each pediatric age range can vary considerably depending on the reagents and analytical equipment employed, and use of published reference ranges is only appropriate where reagents, equipment, and population are identical.[176]
Variations in levels related to age, gender, and acquired conditions, and an overlap of PS levels between mutation carriers and wild-type individuals, can result in misdiagnoses of inherited PS deficiency.[117] Use of lower cutoffs equating to the highest values found in heterozygous carriers rather than the lower limit of a normal population distribution can improve diagnostic specificity for antigen and activity assays,[177] whereas an even lower cutoff for FPS set at <0.10th percentile identifies subjects at risk of VTE.[121]
Activated Protein C Resistance
APCR, defined as a reduced anticoagulant response of plasma to the action of APC, is a prevalent risk factor for VTE.[178] [179] The most common cause of hereditary APCR is FVL (p.Arg534Gln), a single nucleotide polymorphism of F5 that abolishes the predominant APC cleavage site in FVa, Arg506, thereby conferring resistance to APC.[180] APC-mediated cleavage of FV to facilitate its APC-cofactor activity in FVIIIa inactivation occurs at Arg506, further contributing to the hypercoagulability of FVL. FVL is the most common genetic risk factor for VTE, accounting for >90% of hereditary APCR, approximately 20% of unselected patients with first VTE, and up to 50% of patients with familial thrombophilia.[12] [181] Acquired APCR can occur from increases in coagulation factor levels, reductions in inhibitor levels, or presence of autoantibodies, which can arise in a variety of conditions.[181]
Hereditary Activated Protein C Resistance
FVL is an autosomal dominant disorder with variable penetrance, occurring predominantly in Caucasians.[12] [182] Heterozygosity rates are highest in Europe, between 3 and 15% of the populations, whereas it is virtually absent in Asian, African, and native populations of the Americas, Australia, and Greenland.[12] [181] Homozygosity is present in approximately 1 in 5,000 of White populations. Many individuals with FVL never develop VTE, and while most affected individuals do not experience their first event until adulthood, others present with recurrent VTE before 30 years of age. Thrombotic risk is increased when FVL coexists with other risk factors, such as other inherited thrombophilias, elevated FVIII, obesity, oral contraception, HRT, air travel, pregnancy, injury, surgery, and malignancy.[12] Homozygotes tend to develop VTE at a younger age. The phrases FVL and APCR are often used interchangeably, yet several F5 variants have been described since the discovery of FVL that confer different degrees of APCR, through a variety of mechanisms. Some directly affect an APCR cleavage site or make one of the sites inaccessible to APC cleavage, whereas others interfere with FVa binding to negatively charged phospholipids.[181] Variants such as FV Cambridge,[183] FV Hong Kong,[184] FV-Glu666Asp,[185] and FV-Arg485Lys[186] are considered to have weak or negligible thrombotic risk, whereas variants such as FV Liverpool,[187] FV Bonn,[188] FV Nara,[189] FV Kanazawa,[181] and FV Besançon[190] express similar or more marked in vitro APCR to FVL, and increase the risk for VTE.[181]
Acquired Activated Protein C Resistance
Acquired APCR is an independent VTE risk factor and can increase VTE risk in patients with inherited thrombophilia.[181] [191] Elevated coagulation factor levels, particularly of FII and FVIII, are associated with acquired APCR.[181] [192] [193] High FVIII levels can be innate, such as with blood group AB, or circumstantial, such as with stress, exercise, surgery, pregnancy, and other causes of an acute phase response. Markedly elevated FVIII levels associated with severe thrombophilia are encountered with FVIII Padua, a gain-of-function duplication in the FVIII gene.[194] Patients carrying FVIII Padua have normalized APCR ratios (aPTT-based assay) below normal ranges but above that of most FVL heterozygotes (Paulo Simioni, personal communication). In vitro APCR has been reported for FII levels >110 IU/dL, irrespective of whether due to F2 G20210A or not.[181] [195] Acquired APCR can arise from the reduced APC-cofactor activity resulting from deficiencies of FPS.[191]
Reductions in FPS and TFPIα are the main contributors to the hormone-induced APCR that can be encountered in oral contraception, pregnancy, and postmenopausal HRT.[191] [196] The acquired APCR encountered in malignancies is associated with VTE but is also found in cancer patients without thrombosis.[181] In cases of solid tumours, the acquired APCR arises mainly from elevated coagulation factor levels,[197] whereas in hematologic malignancies, it correlates with reduced levels of FPS and TFPIα.[198] Autoantibodies directed to PC, PS, and FV, which are often but not always associated with APS or SLE, can cause acquired APCR and be accompanied by a severe thrombotic phenotype.[181] Additionally, the LAs, anti-β2-glycoprotein I antibodies and anti-phosphatidylserine/prothrombin antibodies found in patients with APS can induce clinically significant acquired APCR.[199]
Phenotypic Detection of Activated Protein C Resistance
Routine phenotypic detection of APCR employs clotting-based assays that assess the degree of anticoagulation achieved by APC.[179] Assays based on thrombin generation/endogenous thrombin potential (ETP) have been described that detect APCR, but they are not in routine use and will not be considered in detail here. The main reason for including these assays in thrombophilia screening is detection or exclusion of FVL, usually with rapid automated assays, yet positive results require confirmatory genetic analysis. Consequently, some centres bypass APCR assays and only perform genetic analysis for FVL, with acceptance that rare F5 variants conferring APCR, and acquired APCR, will go undetected.
Activated Partial Thromboplastin Time-Based Assays
Elegant in the simplicity of its design, the original, classic APCR (CAPCR) assay derives a ratio from the clotting time of a baseline aPTT as denominator, and the same aPTT but in the presence of exogenous APC as numerator.[200] Plasma from patients without APCR achieve significant prolongation of the aPTT in the presence of APC compared with baseline, whereas patients with hereditary APCR and some forms of acquired APCR resist the anticoagulant action of APC, such that the elevation above baseline is less marked, giving rise to a reduced ratio as a marker of APCR. Since the assay is performed on undiluted plasma, other causes of elevated aPTT can falsely elevate ratios and may mask APCR. A good practice is to scrutinize raw data to ensure that the baseline aPTT is normal, to exclude significant interferences, and reject ratios derived from elevated baselines since the additive effect of an existing abnormality or therapeutic anticoagulation on the aPTT + APC can render the ratio unreliable.[179] [201] An exception to this is LAs, because although they may elevate the baseline aPTT, any interference in the phospholipid-dependent PC pathway that shortens the aPTT + APC is a genuine reflection of acquired APCR.[179] [201] [202] The CAPCR assay is sensitive to the acquired APCRs of elevated clotting factors, anti-PC antibodies, and reduced PS if <20% of normal.[179] [181]
Recognition that sensitivity and specificity of CAPCR are reduced, because it is prone to interferences led to development of the modified aPTT-based APCR (MAPCR) assay that is performed identically to CAPCR, except that test plasma is prediluted 1:5 in FV-deficient plasma (FVDP).[179] [200] This modification eliminates interference from clotting factor deficiencies and VKA anticoagulation and corrects for elevated clotting factors and reduced PS. Interference by heparins is negated by the combination of dilution effect and neutralizers contained in most FVDP reagents, although supratherapeutic levels are a potential interference. Interference is possible with DOACs, and plasma pretreatment with charcoal adsorbents has been shown to be effective when testing for APCR in such cases.[45] [138] LAs and anti-PC antibodies may interfere if sufficiently potent to overcome the dilution effect. Baseline raw data scrutiny to exclude significant interferences can also be valuable with MAPCR. Coexistence of FVL and LA is possible in the patient population being tested, and higher plasma dilutions can be used to eliminate the LA effect and permit F5-related APCR to manifest alone.[179] [203] The dilution in FVDP confers almost 100% sensitivity and specificity of MAPCR to F5 mutations conferring APCR, because most interferences are eliminated, and the only significant contribution to the assay reactions from the test plasma is the FV. A modification of CAPCR is available that employs Protac to activate endogenous PC in place of exogenous PC, which can also be used with sample predilution in FVDP.[204]
Snake Venom-Based Assays
Initiation of coagulation with RVV-X, the FX activator from Russell's viper (Daboia russelli) venom, for the baseline clotting time, and addition of Protac to activate endogenous PC for the anticoagulated clotting time, is an alternative to CAPCR.[205] Ratios are generated identically to CAPCR and MAPCR. Bypassing the intrinsic pathway improves specificity, although it negates sensitivity to the acquired APCR of elevated FVIII, whereas high phospholipid concentration reduces LA interference, and the reagents contain a heparin neutralizer. Reduced PC, PS, FII, FV, and FX, and anticoagulation with VKAs and DOACs, can prolong clotting times and falsely elevate ratios. Sensitivity and specificity for F5 variants is improved with sample predilution in FVDP, which corrects for PC, PS, and coagulation factor deficiencies and elevations.[179] [205]
A different approach is taken with an assay employing the FX activator from Southern Pacific rattlesnake (Crotalus viridis helleri) venom in the presence of exogenous APC, phospholipid, and Ca2+.[206] Test plasma is diluted in buffer and then further diluted in FVDP, and clotting times below a reference range are designated as having APCR. Deficiency of FV prolongs clotting times and could mask a coexistent F5 variant conferring APCR, and despite the dilutions, LAs and DOACs are potential interferents if present in high levels.[179] Modification of the assay to generate ratios from clotting times in the presence and absence of APC has been claimed to improve sensitivity and specificity.[207]
Another snake venom assay involves activation of FV by RVV-V, the FV activator from Russell's viper venom, in the presence and absence of exogenous APC.[208] Coagulation is then initiated by Noscarin, a phospholipid- and Ca2+-independent/FV-dependent FII activator from Australian Eastern Tiger snake (Notechis scutatus scutatus) venom. Inactivation of FVa by APC in normal samples leaves little residual FVa to act as cofactor for Noscarin, thereby prolonging clotting times compared with baseline, to generate normal ratios. Plasmas with APC-resistant FV leave more FVa for Noscarin cofactor activity, which shortens clotting times in the presence of APC, resulting in lower ratios. This assay has the highest specificity because dilution in FVDP corrects for factor deficiencies and elevations, and VKA anticoagulation, reagents contain a heparin neutralizer, direct FII activation bypasses direct FXa inhibitor anticoagulants, and the phospholipid-free assay system negates LA interference.[179] Direct thrombin inhibitors, supratherapeutic heparin, and FV deficiency may falsely elevate ratios.[179]
FXa-Based Assay
A mixture of purified fibrinogen, FII, PS, and APC is added to a 1:20 dilution of test plasma in buffer, and coagulation initiated through addition of FXa, phospholipids and Ca2+.[200] [209] Clotting times are read from a calibration curve prepared from dilutions of a pool of plasmas from FVL heterozygotes and converted to percentage FVL. This is of course not entirely accurate because other F5 variants can be expected to read as apparent FVL. The total degree of plasma dilution renders the assay insensitive to most potential interferences and acquired APCR, although FV deficiency will prolong clotting times and potentially mask APCR due to a coexisting F5 variant conferring APCR.
Interpretation of Activated Protein C Resistance Assays
For ratio-based assays, some laboratories normalize results by dividing the patient's ratio by the ratio of a simultaneously analyzed normal pooled plasma (NPP).[179] [200] While this practice reduces variation between reagent batches, it does not improve diagnostic efficacy, and even small numbers of unrealized FVL in the NPP donor population reduces accuracy.[179] [210]
Particularly with predilution assays, there tends to be good separation between ratios of normal plasmas and those with FVL, with some assays also achieving demarcation between heterozygotes and homozygotes.[179] [211] Samples whose ratios are above those realized with FVL plasmas but below the lower limit of the reference range are suggestive of acquired APCR or F5 variants conferring milder APCR than FVL.[179] [201] Use of CAPCR alongside a predilution assay will detect some forms of acquired APCR (i.e., elevated FII or FVIII, severe PS reduction, LA, antibodies to PC, PS, or FV), which can be useful for interpretations when the predilution assay is normal ([Table 4]).[181] [201] ETP-based APCR assays are sensitive to the acquired APCR of reduced FPS, reduced TFPIα, pregnancy/hormone therapy, hematologic malignancies, APS, and other autoantibodies.[181]
|
Interpretation |
CAPCR ratio |
Predilution assay ratio |
|---|---|---|
|
APCR not demonstrated |
Normal |
Normal |
|
Acquired APCR (to which CAPCR is sensitive) |
↓ |
Normal |
|
F5 mutation conferring APCR |
↓ |
↓ |
|
F5 mutation conferring APCR[a] |
Normal |
↓ |
Abbreviations: APCR, activated protein C resistance; aPTT, activated partial thromboplastin time; CAPCR, classic aPTT-based activated protein C resistance assay without sample predilution.
↓: decreased.
a Lower specificity of CAPCR can lead to normal ratios in patients with F5 mutations conferring APCR.[179] [201]
Since APCR testing is performed on plasma, but genetic analysis is undertaken on DNA extracted from white blood cells, genotype/phenotype discrepancies can occur with some liver and bone marrow (BM) transplants. Liver is the site of FV synthesis, so an FVL patient receiving a wild-type liver will achieve phenotypic correction of APCR testing, but genetic analysis will remain positive for FVL.[212] Conversely, the intriguing notion of acquired hereditary APCR occurs in the reverse situation of a wild-type recipient of an FVL liver, where the plasma exhibits newly acquired APCR but DNA analysis for FVL remains normal, which is associated with VTE.[213] [214] The acute phase response associated with liver transplantation surgery can elevate FVIII levels sufficient to generate temporary, acquired APCR.[215] With BM transplants, a wild-type recipient of BM from an FVL donor will generate true negative phenotypic testing, but genetic analysis will convert to FVL positivity, whereas an FVL recipient of a wild-type donor will exhibit true-positive phenotypic testing and genetic analysis will convert to absence of FVL.[216] Genotype–phenotype discordance would also occur for other inherited thrombophilias in these settings, although genotyping is less frequently performed for diagnosis of AT, PC, and PS defects.
Similar to FVL, many of the non-FVL F5 variants achieve APCR through both reduced inactivation of FVa and decreased or absent cofactor activity of FV in FVIIIa inactivation.[181] Since the non-aPTT-based assays bypass FVIIIa inactivation, it seems likely they underestimate the degree of APCR in such cases, although they are sufficiently sensitive to reduced FVa inactivation to be able to detect APCR of such F5 variants. The main APCR mechanism in the FV (H)R2 haplotype is impaired APCR cofactor activity.[181] The markedly reduced FVa inactivation with FV Nara arises from defective interactions with phospholipids and/or PS, which may also explain its absent APC cofactor activity.[181] Thus, it may be that the phospholipid-free RVV-V/Noscarin assay is insensitive to such defects.
Other Thrombophilias
Routine thrombophilia screening should include genetic analysis for F2 G20210A, since there are no reliable phenotypic assays. About 20% of patients with inherited dysfibrinogenemia develop thrombosis, due to defective thrombin binding, defective plasminogen or tissue plasminogen activator binding, or plasmin resistance.[217] At the very least, consideration should be given to inclusion of a thrombin time (TT) in routine thrombophilia screening where an elevated clotting time suggests a fibrinogen abnormality, although the thrombophilic fibrinogen Oslo I generates short TTs due to rapid polymerization.[218] A quantitative Clauss fibrinogen activity assay (not PT-derived fibrinogen) is preferable, as it will generate a reduced result in most cases of inherited dysfibrinogenemia, although between-assay result variability occurs with some variants.[219] A low Clauss fibrinogen can be followed up with fibrinogen antigen where a reduced Clauss–antigen ratio indicates a qualitative abnormality. Since fibrinogen is an acute phase protein, the antigen assay must be performed on the same sample used for the Clauss activity assay. Genetic analysis can confirm the presence and identity of a dysfibrinogen associated with thrombosis.
Numerous other markers of hereditary thrombophilia with unclear clinical significance have been described, such as deficiencies of plasminogen, protein Z, and HCII, and elevations of thrombin activable fibrinolysis inhibitor, plasminogen activator inhibitor-1, and lipoprotein (a). Their assessment is not considered appropriate in routine screening, and they are reviewed elsewhere.[220] [221] The gain of activity thrombophilic variants FVIII Padua[194] and FIX Padua[222] have (so far) only been described in isolated families. Resistance to AT due to mutant FII molecules has been described in families from different countries,[223] [224] [225] [226] and larger studies will inform whether it is sufficiently prevalent for entry into first-line thrombophilia screening. Phenotypic assays have been described for AT resistance, but they are not commercially available.[224] [227]
General Considerations
As with other hemostasis assays, phenotypic thrombophilia testing is affected by a variety of preanalytical variables, such as sample collection, processing, and storage, and postanalytical variables such as interpretation and reporting, requiring knowledge of assay principles and limitations, which are reviewed in detail elsewhere.[228] [229] Calibration plasmas for AT, PC, and PS assays should be traceable to the international standard plasmas for each analyte,[230] quality control should include plasmas at normal and abnormal levels, which should be complemented by regular participation in external quality assessment.[231]
As with FVL, ethnicity is an important consideration when testing for AT, PC, and PS and interpreting results. PS, and to a lesser but significant extent, PC, have been reported to be lower in West Africans than populations of Caucasian and Afro-Caribbean descent,[232] emphasizing the necessity for locally derived reference ranges based on the assays, reagents, and equipment in local use, employing normal donors from the population that will be tested. Higher prevalence of AT, PC, and PS deficiencies have been reported in East Asian countries such as Japan, China, and Taiwan.[233] [234] [235] An interesting type IIb PC variant prevalent in the Chinese population (p.Lys193del) and absent in Caucasians returns normal results in chromogenic PC activity assays, and whereas PC activity measured with a clotting-based activity is lower, it may also be within the reference range.[236]
Aside from the debate about who to test, “when to test” (with phenotypic assays) is an important consideration. Phenotypic screening should not be undertaken in the acute phase after the thromboembolic event due to potential for consumption of physiological anticoagulants.[2] It should be performed at least 3 months after the VTE, and additionally, at least 2 weeks after discontinuing VKA therapy, or 2 days for DOACs and heparins, although some advocate longer periods.[2] [15] PC and PS are reduced on VKA therapy, so results cannot reliably differentiate between normality and hereditary deficiency, and all other anticoagulants are potential interferents in at least some assays unless assay design such as sample dilution and reagent-integral heparin neutralizers are effective, or preanalytics like charcoal adsorbents are employed. However, these strategies have their own limitations, with which practitioners involved in performing and interpreting test results must be fully familiar. Phenotypic thrombophilia testing should not be undertaken during pregnancy or immediately postpartum due to gestational hemostatic alterations, which begin at conception and can be present up to 12 weeks postnatally.[237] Similarly, oral contraception and HRT can affect thrombophilia screening results, which must be borne in mind during interpretation if the treatment cannot be interrupted. Age of the patient must also be considered when interpreting results. It is commonly advised to repeat analysis of any parameters generating abnormal results on a fresh sample, to account for issues such as physiological variation, natural statistical outliers, preanalytical considerations, and analytical variations, yet logically this should extend to normal results that may be abnormal on repeat testing.
Minimizing risk of assay interferences and misinterpretations is a shared responsibility; for clinicians in avoiding requests for testing when assays may be compromised, or supplying all relevant information to permit informed assay performance and interpretation, or rejection; for laboratory scientists to take appropriate precautions, up to and including refusal to perform a given test; and scientists or clinicians undertaking interpretive reporting to do so with maximum patient information, knowledge of physiology and pathophysiology, and awareness of assay limitations.
Conclusion
No single phenotypic assay will detect every clinically significant deficiency of AT, PC, and PS, and negative testing does not necessarily mean a patient is free of thrombophilia. Where identification of a thrombophilia is considered likely and useful to management, laboratories should consider availability of alternative assays to those in local routine use to widen the diagnostic net, be that in-house or referral to other centres where appropriate assays are available. Ultimately, genetic analysis may be complementary or necessary where phenotypic testing is compromised or equivocal.
Conflict of Interest
G.W.M. reports consultancy fees from Technoclone.
-
References
- 1 Colucci G, Tsakiris DA. Thrombophilia screening revisited: an issue of personalized medicine. J Thromb Thrombolysis 2020; 49 (04) 618-629
- 2 Colucci G, Tsakiris DA. Thrombophilia screening: universal, selected, or neither?. Clin Appl Thromb Hemost 2017; 23 (08) 893-899
- 3 Srivastava A, Brewer AK, Mauser-Bunschoten EP. et al; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19 (01) e1-e47
- 4 Heit JA. Thrombophilia: common questions on laboratory assessment and management. Hematology (Am Soc Hematol Educ Program) 2007; 2007: 127-135
- 5 Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood 2008; 112 (01) 19-27
- 6 Zöller B, Svensson PJ, Dahlbäck B, Lind-Hallden C, Hallden C, Elf J. Genetic risk factors for venous thromboembolism. Expert Rev Hematol 2020; 13 (09) 971-981
- 7 Arachchillage DJ, Mackillop L, Chandratheva A, Motawani J, MacCallum P, Laffan M. Thrombophilia testing: a British Society for Haematology guideline. Br J Haematol 2022; 198 (03) 443-458
- 8 Mannucci PM, Franchini M. Classic thrombophilic gene variants. Thromb Haemost 2015; 114 (05) 885-889
- 9 Van Cott EM, Khor B, Zehnder JL, Factor V. Factor V Leiden. Am J Hematol 2016; 91 (01) 46-49
- 10 Rolla R, Pergolini P, Vidali M. et al. Routine coagulation tests are not useful as a screening tool for the FII G20210A polymorphism. Clin Lab 2014; 60 (10) 1725-1733
- 11 Nicolaes GA, Dahlbäck B. Activated protein C resistance (FV(Leiden)) and thrombosis: factor V mutations causing hypercoagulable states. Hematol Oncol Clin North Am 2003; 17 (01) 37-61 , vi
- 12 Kujovich JL, Factor V. Factor V Leiden thrombophilia. Genet Med 2011; 13 (01) 1-16
- 13 Middeldorp S, Nieuwlaat R, Baumann Kreuziger L. et al. American Society of Hematology 2023 guidelines for management of venous thromboembolism: thrombophilia testing. Blood Adv 2023; 7 (22) 7101-7138
- 14 Kudo M, Lee HL, Yang IA, Masel PJ. Utility of thrombophilia testing in patients with venous thrombo-embolism. J Thorac Dis 2016; 8 (12) 3697-3703
- 15 Connors JM. Thrombophilia testing and venous thrombosis. N Engl J Med 2017; 377 (12) 1177-1187
- 16 De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110 (04) 697-705
- 17 Stevens SM, Woller SC, Bauer KA. et al. Guidance for the evaluation and treatment of hereditary and acquired thrombophilia. J Thromb Thrombolysis 2016; 41 (01) 154-164
- 18 MacCallum P, Bowles L, Keeling D. Diagnosis and management of heritable thrombophilias. BMJ 2014; 349: g4387
- 19 Middeldorp S. Inherited thrombophilia: a double-edged sword. Hematology (Am Soc Hematol Educ Program) 2016; 2016 (01) 1-9
- 20 Franchini M. The utility of thrombophilia testing. Clin Chem Lab Med 2014; 52 (04) 495-497
- 21 Fell C, Ivanovic N, Johnson SA, Seegers WH. Differentiation of plasma antithrombin activities. Proc Soc Exp Biol Med 1954; 85 (02) 199-202
- 22 Kottke-Marchant K, Duncan A. Antithrombin deficiency: issues in laboratory diagnosis. Arch Pathol Lab Med 2002; 126 (11) 1326-1336
- 23 Rezaie AR, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost 2020; 18 (12) 3142-3153
- 24 Muszbek L, Bereczky Z, Kovács B, Komáromi I. Antithrombin deficiency and its laboratory diagnosis. Clin Chem Lab Med 2010; 48 (Suppl. 01) S67-S78
- 25 Pabinger I, Thaler J. How I treat patients with hereditary antithrombin deficiency. Blood 2019; 134 (26) 2346-2353
- 26 Law RH, Zhang Q, McGowan S. et al. An overview of the serpin superfamily. Genome Biol 2006; 7 (05) 216
- 27 Opal SM, Kessler CM, Roemisch J, Knaub S. Antithrombin, heparin, and heparan sulfate. Crit Care Med 2002; 30 (5, Suppl): S325-S331
- 28 Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci U S A 1997; 94 (26) 14683-14688
- 29 Liu J, Pedersen LC. Anticoagulant heparan sulfate: structural specificity and biosynthesis. Appl Microbiol Biotechnol 2007; 74 (02) 263-272
- 30 Swedenborg J. The mechanisms of action of alpha- and beta-isoforms of antithrombin. Blood Coagul Fibrinolysis 1998; 9 (Suppl. 03) S7-S10
- 31 Teitel JM, Rosenberg RD. Protection of factor Xa from neutralization by the heparin-antithrombin complex. J Clin Invest 1983; 71 (05) 1383-1391
- 32 Weitz JI. Heparan sulfate: antithrombotic or not?. J Clin Invest 2003; 111 (07) 952-954
- 33 Egeberg O. Thrombophilia caused by inheritable deficiency of blood antithrombin. Scand J Clin Lab Invest 1965; 17: 92
- 34 Bravo-Pérez C, de la Morena-Barrio ME, Vicente V, Corral J. Antithrombin deficiency as a still underdiagnosed thrombophilia: a primer for internists. Pol Arch Intern Med 2020; 130 (10) 868-877
- 35 Alhenc-Gelas M, Plu-Bureau G, Hugon-Rodin J, Picard V, Horellou MH. GFHT study group on Genetic Thrombophilia. Thrombotic risk according to SERPINC1 genotype in a large cohort of subjects with antithrombin inherited deficiency. Thromb Haemost 2017; 117 (06) 1040-1051
- 36 Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia 2008; 14 (06) 1229-1239
- 37 Corral J, de la Morena-Barrio ME, Vicente V. The genetics of antithrombin. Thromb Res 2018; 169: 23-29
- 38 Brown SA, Mitchell M, Cutler JA, Moore G, Smith MP, Savidge GF. Rapid genetic diagnosis in neonatal pulmonary artery thrombosis caused by homozygous antithrombin Budapest 3. Clin Appl Thromb Hemost 2000; 6 (03) 181-183
- 39 Van Cott EM, Orlando C, Moore GW, Cooper PC, Meijer P, Marlar R. Subcommittee on Plasma Coagulation Inhibitors. Recommendations for clinical laboratory testing for antithrombin deficiency; communication from the SSC of the ISTH. J Thromb Haemost 2020; 18 (01) 17-22
- 40 Cooper PC, Coath F, Daly ME, Makris M. The phenotypic and genetic assessment of antithrombin deficiency. Int J Lab Hematol 2011; 33 (03) 227-237
- 41 Kovács B, Bereczky Z, Oláh Z. et al. The superiority of anti-FXa assay over anti-FIIa assay in detecting heparin-binding site antithrombin deficiency. Am J Clin Pathol 2013; 140 (05) 675-679
- 42 Moser KA, Smock KJ. Direct oral anticoagulant (DOAC) interference in hemostasis assays. Hematology (Am Soc Hematol Educ Program) 2021; 2021 (01) 129-133
- 43 Smogorzewska A, Brandt JT, Chandler WL. et al. Effect of fondaparinux on coagulation assays: results of College of American Pathologists proficiency testing. Arch Pathol Lab Med 2006; 130 (11) 1605-1611
- 44 Van Blerk M, Bailleul E, Chatelain B. et al. Influence of dabigatran and rivaroxaban on routine coagulation assays. A nationwide Belgian survey. Thromb Haemost 2015; 113 (01) 154-164
- 45 Favresse J, Lardinois B, Sabor L. et al. Evaluation of the DOAC-Stop® procedure to overcome the effect of DOACs on several thrombophilia screening tests. TH Open 2018; 2 (02) e202-e209
- 46 Antovic J, Söderström J, Karlman B, Blombäck M. Evaluation of a new immunoturbidimetric test (Liatest antithrombin III) for determination of antithrombin antigen. Clin Lab Haematol 2001; 23 (05) 313-316
- 47 Heit JA. Thrombophilia: clinical and laboratory assessment and management. In: Kitchens CS, Alving BM, Kessler CM. eds. Consultative Hemostasis and Thrombosis. 2nd ed.. Philadelphia: Saunders; 2007: 213-244
- 48 Kovács B, Bereczky Z, Selmeczi A. et al. Progressive chromogenic anti-factor Xa assay and its use in the classification of antithrombin deficiencies. Clin Chem Lab Med 2014; 52 (12) 1797-1806
- 49 Chowdhury V, Mille B, Olds RJ. et al. Antithrombins Southport (Leu 99 to Val) and Vienna (Gln 118 to Pro): two novel antithrombin variants with abnormal heparin binding. Br J Haematol 1995; 89 (03) 602-609
- 50 Moore GW, de Jager N, Cutler JA. Development of a novel, rapid assay for detection of heparin-binding defect antithrombin deficiencies: the heparin-antithrombin binding (HAB) ratio. Thromb Res 2015; 135 (01) 161-166
- 51 Corral J, Hernandez-Espinosa D, Soria JM. et al. Antithrombin Cambridge II (A384S): an underestimated genetic risk factor for venous thrombosis. Blood 2007; 109 (10) 4258-4263
- 52 Perry DJ, Daly ME, Tait RC. et al. Antithrombin Cambridge II (Ala384Ser): clinical, functional and haplotype analysis of 18 families. Thromb Haemost 1998; 79 (02) 249-253
- 53 Olson ST, Stephens AW, Hirs CH, Bock PE, Björk I. Kinetic characterization of the proteinase binding defect in a reactive site variant of the serpin, antithrombin. Role of the P1′ residue in transition-state stabilization of antithrombin-proteinase complex formation. J Biol Chem 1995; 270 (17) 9717-9724
- 54 Ungerstedt JS, Schulman S, Egberg N, Antovic J, Blombäck N. Discrepancy between antithrombin activity methods revealed in antithrombin Stockholm: do factor Xa-based methods overestimate antithrombin activity in some patients?. Blood 2002; 99 (06) 2271-2272
- 55 Lane DA, Lowe GD, Flynn A, Thompson E, Ireland H, Erdjument H. Antithrombin III Glasgow: a variant with increased heparin affinity and reduced ability to inactivate thrombin, associated with familial thrombosis. Br J Haematol 1987; 66 (04) 523-527
- 56 Beauchamp NJ, Pike RN, Daly M. et al. Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood 1998; 92 (08) 2696-2706
- 57 Bravo-Pérez C, de la Morena-Barrio ME, de la Morena-Barrio B. et al. Molecular and clinical characterization of transient antithrombin deficiency: a new concept in congenital thrombophilia. Am J Hematol 2022; 97 (02) 216-225
- 58 Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn–>Asp). J Clin Invest 1994; 94 (06) 2265-2274
- 59 Picard V, Bauters A, Khairy M. et al. Conformational Asn187Asp/Lys antithrombin variants and thrombosis. Clinical and biological features in 13 new heterozygotes. Thromb Haemost 2005; 93 (01) 57-62
- 60 Navarro-Fernández J, de la Morena-Barrio ME, Padilla J. et al. Antithrombin Dublin (p.Val30Glu): a relatively common variant with moderate thrombosis risk of causing transient antithrombin deficiency. Thromb Haemost 2016; 116 (01) 146-154
- 61 Weronska A, De la Morena-Barrio B, Goldman-Mazur S. et al. Functional, biochemical, molecular and clinical characterization of antithrombin c.1157T>C (p.Ile386Thr), a recurrent Polish variant with a founder effect. Haematologica 2023; 108 (10) 2803-2807
- 62 de la Morena-Barrio ME, Suchon P, Jacobsen EM. et al. Two SERPINC1 variants affecting N-glycosylation of Asn224 cause severe thrombophilia not detected by functional assays. Blood 2022; 140 (02) 140-151
- 63 Goldenberg NA, Manco-Johnson MJ. Protein C deficiency. Haemophilia 2008; 14 (06) 1214-1221
- 64 Stenflo J. A new vitamin K-dependent protein. Purification from bovine plasma and preliminary characterization. J Biol Chem 1976; 251 (02) 355-363
- 65 Dahlbäck B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol 2005; 25 (07) 1311-1320
- 66 Thiyagarajan M, Cheng T, Zlokovic BV. Endothelial cell protein C receptor: role beyond endothelium?. Circ Res 2007; 100 (02) 155-157
- 67 Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018; 132 (02) 148-158
- 68 Kottke-Marchant K, Comp P. Laboratory issues in diagnosing abnormalities of protein C, thrombomodulin, and endothelial cell protein C receptor. Arch Pathol Lab Med 2002; 126 (11) 1337-1348
- 69 Nicolaes GA, Dahlbäck B. Factor V and thrombotic disease: description of a Janus-faced protein. Arterioscler Thromb Vasc Biol 2002; 22 (04) 530-538
- 70 Dinarvand P, Moser KA, Protein C. Deficiency. Arch Pathol Lab Med 2019; 143 (10) 1281-1285
- 71 Bahou WF. Thrombin's faces revealed: cellular effects include induction of neoangiogenesis. J Thromb Haemost 2003; 1 (10) 2078-2080
- 72 MacMahon A. The realm of Janus: Doorways in the Roman World. In: Carr G, Swift E, Weekes J. eds. TRAC 2002: Proceedings of the Twelfth Annual Theoretical Roman Archaeology Conference, Canterbury 2002. Oxford: Oxbow Books; 2003: 58-73
- 73 Suzuki K. The multi-functional serpin, protein C inhibitor: beyond thrombosis and hemostasis. J Thromb Haemost 2008; 6 (12) 2017-2026
- 74 Marlar RA, Gausman JN. Laboratory testing issues for protein C, protein S, and antithrombin. Int J Lab Hematol 2014; 36 (03) 289-295
- 75 Minford A, Brandão LR, Othman M. et al. Diagnosis and management of severe congenital protein C deficiency (SCPCD): communication from the SSC of the ISTH. J Thromb Haemost 2022; 20 (07) 1735-1743
- 76 Tripodi A, Franchi F, Krachmalnicoff A, Mannucci PM. Asymptomatic homozygous protein C deficiency. Acta Haematol 1990; 83 (03) 152-155
- 77 Shakoor MT, Ayub S, Ayub Z. Warfarin induced skin necrosis: a diagnostic challenge. MOJ Clin Med Case Rep 2017; 6: 21-22
- 78 Khor B, Van Cott EM. Laboratory tests for protein C deficiency. Am J Hematol 2010; 85 (06) 440-442
- 79 Cooper PC, Pavlova A, Moore GW, Hickey KP, Marlar RA. Recommendations for clinical laboratory testing for protein C deficiency, for the subcommittee on plasma coagulation inhibitors of the ISTH. J Thromb Haemost 2020; 18 (02) 271-277
- 80 Moore GW. Snake venoms in diagnostic hemostasis and thrombosis. Semin Thromb Hemost 2022; 48 (02) 145-160
- 81 Cooper PC, Hill M, Maclean RM. The phenotypic and genetic assessment of protein C deficiency. Int J Lab Hematol 2012; 34 (04) 336-346
- 82 Simioni P, Lazzaro A, Zanardi S, Girolami A. Spurious protein C deficiency due to antiphospholipid antibodies. Am J Hematol 1991; 36 (04) 299-301
- 83 Favre R, Zia-Chahabi S, Talb Y, de Gunzburg N, Flaujac C. Direct oral anticoagulant neutralization by activated charcoal DOAC-remove for thrombophilia screening. Blood Coagul Fibrinolysis 2021; 32 (05) 356-358
- 84 Ruinemans-Koerts J, Peterse-Stienissen I, Verbruggen B. Non-parallelism in the one-stage coagulation factor assay is a phenomenon of lupus anticoagulants and not of individual factor inhibitors. Thromb Haemost 2010; 104 (05) 1080-1082
- 85 Mackie IJ, Gallimore M, Machin SJ. Contact factor proteases and the complexes formed with alpha 2-macroglobulin can interfere in protein C assays by cleaving amidolytic substrates. Blood Coagul Fibrinolysis 1992; 3 (05) 589-595
- 86 Han P, Fung KP, Rahdakrishnan U. Lack of agreement of chromogenic and clotting assays for factor X and protein C in warfarinised plasma. Thromb Haemost 1991; 65 (04) 360-363
- 87 Bovill EG, Tomczak JA, Grant B. et al. Protein CVermont: symptomatic type II protein C deficiency associated with two GLA domain mutations. Blood 1992; 79 (06) 1456-1465
- 88 Gaussem P, Gandrille S, Duchemin J. et al. Influence of six mutations of the protein C gene on the Gla domain conformation and calcium affinity. Thromb Haemost 1994; 71 (06) 748-754
- 89 Seidel H, Haracska B, Naumann J, Westhofen P, Hass MS, Kruppenbacher JP. Laboratory limitations of excluding hereditary protein C deficiency by chromogenic assay: discrepancies of phenotype and genotype. Clin Appl Thromb Hemost 2020; 26: 1076029620912028
- 90 Cooper PC, Siddiq S, Morse C, Nightingale J, Mumford AD. Marked discrepancy between coagulometric protein C activity assays with the pro-thrombotic protein C Asn2Ile substitution. Int J Lab Hematol 2011; 33 (05) 451-456
- 91 Noguchi K, Nakazono E, Tsuda T. et al. Plasma phenotypes of protein S Lys196Glu and protein C Lys193del variants prevalent among young Japanese women. Blood Coagul Fibrinolysis 2019; 30 (08) 393-400
- 92 Efthymiou M, Arachchillage DRJ, Lane PJ. et al. Antibodies against TFPI and protein C are associated with a severe thrombotic phenotype in patients with and without antiphospholipid syndrome. Thromb Res 2018; 170: 60-68
- 93 MacCallum PK, Cooper JA, Martin J, Howarth DJ, Meade TW, Miller GJ. Associations of protein C and protein S with serum lipid concentrations. Br J Haematol 1998; 102 (02) 609-615
- 94 Cushman M, Psaty BM, Meilahn EN, Dobs AS, Kuller LH. Post-menopausal hormone therapy and concentrations of protein C and antithrombin in elderly women. Br J Haematol 2001; 114 (01) 162-168
- 95 Said JM, Ignjatovic V, Monagle PT, Walker SP, Higgins JR, Brennecke SP. Altered reference ranges for protein C and protein S during early pregnancy: implications for the diagnosis of protein C and protein S deficiency during pregnancy. Thromb Haemost 2010; 103 (05) 984-988
- 96 Monagle P, Barnes C, Ignjatovic V. et al. Developmental haemostasis. Impact for clinical haemostasis laboratories. Thromb Haemost 2006; 95 (02) 362-372
- 97 Toulon P. Developmental hemostasis: laboratory and clinical implications. Int J Lab Hematol 2016; 38 (Suppl. 01) 66-77
- 98 Appel IM, Grimminck B, Geerts J, Stigter R, Cnossen MH, Beishuizen A. Age dependency of coagulation parameters during childhood and puberty. J Thromb Haemost 2012; 10 (11) 2254-2263
- 99 Andrew M, Paes B, Milner R. et al. Development of the human coagulation system in the full-term infant. Blood 1987; 70 (01) 165-172
- 100 Andrew M, Vegh P, Johnston M, Bowker J, Ofosu F, Mitchell L. Maturation of the hemostatic system during childhood. Blood 1992; 80 (08) 1998-2005
- 101 Toulon P, Berruyer M, Brionne-François M. et al. Age dependency for coagulation parameters in paediatric populations. Results of a multicentre study aimed at defining the age-specific reference ranges. Thromb Haemost 2016; 116 (01) 9-16
- 102 Attard C, van der Straaten T, Karlaftis V, Monagle P, Ignjatovic V. Developmental hemostasis: age-specific differences in the levels of hemostatic proteins. J Thromb Haemost 2013; 11 (10) 1850-1854
- 103 Conlan MG, Folsom AR, Finch A, Davis CE, Sorlie P, Wu KK. Correlation of plasma protein C levels with cardiovascular risk factors in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) study. Thromb Haemost 1993; 70 (05) 762-767
- 104 Tait RC, Walker ID, Islam SI. et al. Protein C activity in healthy volunteers–influence of age, sex, smoking and oral contraceptives. Thromb Haemost 1993; 70 (02) 281-285
- 105 Lowe GD, Rumley A, Woodward M. et al. Epidemiology of coagulation factors, inhibitors and activation markers: the Third Glasgow MONICA Survey. I. Illustrative reference ranges by age, sex and hormone use. Br J Haematol 1997; 97 (04) 775-784
- 106 Franchi F, Biguzzi E, Martinelli I. et al. Normal reference ranges of antithrombin, protein C and protein S: effect of sex, age and hormonal status. Thromb Res 2013; 132 (02) e152-e157
- 107 Rodeghiero F, Tosetto A. The VITA Project: population-based distributions of protein C, antithrombin III, heparin-cofactor II and plasminogen–relationship with physiological variables and establishment of reference ranges. Thromb Haemost 1996; 76 (02) 226-233
- 108 Allaart CF, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briët E. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet 1993; 341 (8838): 134-138
- 109 Di Scipio RG, Hermodson MA, Yates SG, Davie EW. A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry 1977; 16 (04) 698-706
- 110 Ahnström J, Andersson HM, Canis K. et al. Activated protein C cofactor function of protein S: a novel role for a γ-carboxyglutamic acid residue. Blood 2011; 117 (24) 6685-6693
- 111 Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost 2009; 7 (Suppl. 01) 165-168
- 112 Gierula M, Ahnström J. Anticoagulant protein S-New insights on interactions and functions. J Thromb Haemost 2020; 18 (11) 2801-2811
- 113 Dahlbäck B, Guo LJ, Livaja-Koshiar R, Tran S. Factor V-short and protein S as synergistic tissue factor pathway inhibitor (TFPIα) cofactors. Res Pract Thromb Haemost 2017; 2 (01) 114-124
- 114 Gierula M, Noakes VM, Salles-Crawley II, Crawley JTB, Ahnström J. The TFPIα C-terminal tail is essential for TFPIα-FV-short-protein S complex formation and synergistic enhancement of TFPIα. J Thromb Haemost 2023; 21 (12) 3568-3580
- 115 Dahlbäck B. The tale of protein S and C4b-binding protein, a story of affection. Thromb Haemost 2007; 98 (01) 90-96
- 116 Maurissen LF, Thomassen MC, Nicolaes GA. et al. Re-evaluation of the role of the protein S-C4b binding protein complex in activated protein C-catalyzed factor Va-inactivation. Blood 2008; 111 (06) 3034-3041
- 117 ten Kate MK, van der Meer J. Protein S deficiency: a clinical perspective. Haemophilia 2008; 14 (06) 1222-1228
- 118 Marlar RA, Gausman JN, Tsuda H, Rollins-Raval MA, Brinkman HJM. Recommendations for clinical laboratory testing for protein S deficiency: communication from the SSC committee plasma coagulation inhibitors of the ISTH. J Thromb Haemost 2021; 19 (01) 68-74
- 119 Zöller B, García de Frutos P, Dahlbäck B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood 1995; 85 (12) 3524-3531
- 120 Marlar RA, Gausman JN. Protein S abnormalities: a diagnostic nightmare. Am J Hematol 2011; 86 (05) 418-421
- 121 Pintao MC, Ribeiro DD, Bezemer ID. et al. Protein S levels and the risk of venous thrombosis: results from the MEGA case-control study. Blood 2013; 122 (18) 3210-3219
- 122 Anderson DR, Brill-Edwards P, Walker I. Warfarin-induced skin necrosis in 2 patients with protein S deficiency: successful reinstatement of warfarin therapy. Haemostasis 1992; 22 (03) 124-128
- 123 Tai CY, Ierardi R, Alexander JB. A case of warfarin skin necrosis despite enoxaparin anticoagulation in a patient with protein S deficiency. Ann Vasc Surg 2004; 18 (02) 237-242
- 124 Fraga R, Diniz LM, Lucas EA, Emerich PS. Warfarin-induced skin necrosis in a patient with protein S deficiency. An Bras Dermatol 2018; 93 (04) 612-613
- 125 Tsuda T, Jin X, Tsuda H. et al. New quantitative total protein S-assay system for diagnosing protein S type II deficiency: clinical application of the screening system for protein S type II deficiency. Blood Coagul Fibrinolysis 2012; 23 (01) 56-63
- 126 Goodwin AJ, Rosendaal FR, Kottke-Marchant K, Bovill EG. A review of the technical, diagnostic, and epidemiologic considerations for protein S assays. Arch Pathol Lab Med 2002; 126 (11) 1349-1366
- 127 Smock KJ, Plumhoff EA, Meijer P. et al. Protein S testing in patients with protein S deficiency, factor V Leiden, and rivaroxaban by North American Specialized Coagulation Laboratories. Thromb Haemost 2016; 116 (01) 50-57
- 128 Lawrie AS, Lloyd ME, Mohamed F, Irons S, Hughes GR, Savidge GF. Assay of protein S in systemic lupus erythematosus. Blood Coagul Fibrinolysis 1995; 6 (04) 322-324
- 129 Rossi E, Gatti L, Guarneri D, Finotto E, Lombardi A, Preda L. Functional protein S in women with lupus anticoagulant inhibitor. Thromb Res 1992; 65 (02) 253-262
- 130 Tomás JF, Alberca I, Tabernero MD, Cordero M, Del Pino-Montes J, Vicente V. Natural anticoagulant proteins and antiphospholipid antibodies in systemic lupus erythematosus. J Rheumatol 1998; 25 (01) 57-62
- 131 Seriolo B, Cutolo M, Accardo S. Association between acquired free protein S deficiency, anticardiolipin antibodies, and thrombotic events in rheumatoid arthritis. J Rheumatol 1998; 25 (11) 2281-2282
- 132 Parke AL, Weinstein RE, Bona RD, Maier DB, Walker FJ. The thrombotic diathesis associated with the presence of phospholipid antibodies may be due to low levels of free protein S. Am J Med 1992; 93 (01) 49-56
- 133 Van Cott EM, Ledford-Kraemer M, Meijer P, Nichols WL, Johnson SM, Peerschke EI. NASCOLA Proficiency Testing Committee. Protein S assays: an analysis of North American Specialized Coagulation Laboratory Association proficiency testing. Am J Clin Pathol 2005; 123 (05) 778-785
- 134 Jennings I, Kitchen S, Woods TA, Preston FE. Multilaboratory testing in thrombophilia through the United Kingdom National External Quality Assessment Scheme (Blood Coagulation) Quality Assurance Program. Semin Thromb Hemost 2005; 31 (01) 66-72
- 135 Jennings I, Kitchen S, Cooper P, Makris M, Preston FE. Sensitivity of functional protein S assays to protein S deficiency: a comparative study of three commercial kits. J Thromb Haemost 2003; 1 (05) 1112-1114
- 136 Marlar RA, Potts RM, Welsh CW. Accuracy of diagnosis of protein S deficiency by protein S activity and antigen assays. J Clin Ligand 2005; 28: 130-135
- 137 Rodger MA, Carrier M, Gervais M, Rock G. Normal functional protein S activity does not exclude protein S deficiency. Pathophysiol Haemost Thromb 2003; 33 (04) 202-205
- 138 Siriez R, Dogné JM, Gosselin R, Laloy J, Mullier F, Douxfils J. Comprehensive review of the impact of direct oral anticoagulants on thrombophilia diagnostic tests: practical recommendations for the laboratory. Int J Lab Hematol 2021; 43 (01) 7-20
- 139 Bovill EG, Landesman MM, Busch SA, Fregeau GR, Mann KG, Tracy RP. Studies on the measurement of protein S in plasma. Clin Chem 1991; 37 (10 Pt 1): 1708-1714
- 140 Hubbard AR, Wong MY. A collaborative study on the measurement of protein S antigen in plasma. Thromb Haemost 1992; 68 (02) 115-118
- 141 Persson KE, Hillarp A, Dahlbäck B. Analytical considerations for free protein S assays in protein S deficiency. Thromb Haemost 2001; 86 (05) 1144-1147
- 142 Duebgen S, Kauke T, Marschall C. et al. Genotype and laboratory and clinical phenotypes of protein s deficiency. Am J Clin Pathol 2012; 137 (02) 178-184
- 143 Brinkman HJM, Ahnström J, Castoldi E, Dahlbäck B, Marlar RA. Pleiotropic anticoagulant functions of protein S, consequences for the clinical laboratory. Communication from the SSC of the ISTH. J Thromb Haemost 2021; 19 (01) 281-286
- 144 Bertina RM, Ploos van Amstel HK, van Wijngaarden A. et al. Heerlen polymorphism of protein S, an immunologic polymorphism due to dimorphism of residue 460. Blood 1990; 76 (03) 538-548
- 145 Duchemin J, Gandrille S, Borgel D. et al. The Ser 460 to Pro substitution of the protein S alpha (PROS1) gene is a frequent mutation associated with free protein S (type IIa) deficiency. Blood 1995; 86 (09) 3436-3443
- 146 Varvenne M, Kochhan L, Trummer A, Eisert R, Birschmann I. Clinical consequences of compound heterozygosity for protein S mutation Heerlen and p.Cys252Gly protein S mutation. Thromb Res 2011; 128 (05) 498-500
- 147 Wypasek E, Potaczek DP, Alhenc-Gelas M, Undas A. Heerlen polymorphism associated with type III protein S deficiency and factor V Leiden mutation in a Polish patient with deep vein thrombosis. Blood Coagul Fibrinolysis 2014; 25 (01) 84-85
- 148 Suchon P, Germain M, Delluc A. et al. Protein S Heerlen mutation heterozygosity is associated with venous thrombosis risk. Sci Rep 2017; 7: 45507
- 149 Espinosa-Parrilla Y, Navarro G, Morell M, Abella E, Estivill X, Sala N. Homozygosity for the protein S Heerlen allele is associated with type I PS deficiency in a thrombophilic pedigree with multiple risk factors. Thromb Haemost 2000; 83 (01) 102-106
- 150 Alhenc-Gelas M, Canonico M, Morange PE, Emmerich J. Geht Genetic Thrombophilia Group. Protein S inherited qualitative deficiency: novel mutations and phenotypic influence. J Thromb Haemost 2010; 8 (12) 2718-2726
- 151 Denis CV, Roberts SJ, Hackeng TM, Lenting PJ. In vivo clearance of human protein S in a mouse model: influence of C4b-binding protein and the Heerlen polymorphism. Arterioscler Thromb Vasc Biol 2005; 25 (10) 2209-2215
- 152 Heeb MJ, Koenen RR, Fernández JA, Hackeng TM. Direct anticoagulant activity of protein S-C4b binding protein complex in Heerlen heterozygotes and normals. J Thromb Haemost 2004; 2 (10) 1766-1773
- 153 Hayashi T, Nishioka J, Suzuki K. Molecular mechanism of the dysfunction of protein S(Tokushima) (Lys155–>Glu) for the regulation of the blood coagulation system. Biochim Biophys Acta 1995; 1272 (03) 159-167
- 154 D'Angelo A, Viganò D'Angelo S. Protein S deficiency. Haematologica 2008; 93 (04) 498-501
- 155 Vigano-D'Angelo S, D'Angelo A, Kaufman Jr CE, Sholer C, Esmon CT, Comp PC. Protein S deficiency occurs in the nephrotic syndrome. Ann Intern Med 1987; 107 (01) 42-47
- 156 Ceriello A, Giugliano D, Quatraro A, Marchi E, Barbanti M, Lefebvre P. Possible role for increased C4b-binding-protein level in acquired protein S deficiency in type I diabetes. Diabetes 1990; 39 (04) 447-449
- 157 Addai-Mensah O, Annani-Akollor ME, Nsafoah FO. et al. Effect of poor glycaemic control on plasma levels and activity of protein C, protein S, and antithrombin III in type 2 diabetes mellitus. PLoS One 2019; 14 (09) e0223171
- 158 Fernández JA, Estellés A, Gilabert J, España F, Aznar J. Functional and immunologic protein S in normal pregnant women and in full-term newborns. Thromb Haemost 1989; 61 (03) 474-478
- 159 Fu M, Liu J, Xing J. et al. Reference intervals for coagulation parameters in non-pregnant and pregnant women. Sci Rep 2022; 12 (01) 1519
- 160 Szecsi PB, Jørgensen M, Klajnbard A, Andersen MR, Colov NP, Stender S. Haemostatic reference intervals in pregnancy. Thromb Haemost 2010; 103 (04) 718-727
- 161 Oruç S, Saruç M, Koyuncu FM, Ozdemir E. Changes in the plasma activities of protein C and protein S during pregnancy. Aust N Z J Obstet Gynaecol 2000; 40 (04) 448-450
- 162 Stahl CP, Wideman CS, Spira TJ, Haff EC, Hixon GJ, Evatt BL. Protein S deficiency in men with long-term human immunodeficiency virus infection. Blood 1993; 81 (07) 1801-1807
- 163 Lafeuillade A, Sorice M, Griggi T, Pellegrino P, Geoffroy M, Profizi N. Role of autoimmunity in protein S deficiency during HIV-1 infection. Infection 1994; 22 (03) 201-203
- 164 D'Angelo A, Della Valle P, Crippa L, Pattarini E, Grimaldi LM, Viganò D'Angelo S. Brief report: autoimmune protein S deficiency in a boy with severe thromboembolic disease. N Engl J Med 1993; 328 (24) 1753-1757
- 165 Regnault V, Boehlen F, Ozsahin H. et al. Anti-protein S antibodies following a varicella infection: detection, characterization and influence on thrombin generation. J Thromb Haemost 2005; 3 (06) 1243-1249
- 166 Deitcher SR, Erban JK, Limentani SA. Acquired free protein S deficiency associated with multiple myeloma: a case report. Am J Hematol 1996; 51 (04) 319-323
- 167 Guermazi S, Hamza M, Dellagi K. Protein S deficiency and antibodies to protein S in patients with Behçet's disease. Thromb Res 1997; 86 (03) 197-204
- 168 Crowther MA, Johnston M, Weitz J, Ginsberg JS. Free protein S deficiency may be found in patients with antiphospholipid antibodies who do not have systemic lupus erythematosus. Thromb Haemost 1996; 76 (05) 689-691
- 169 Erkan D, Zhang HW, Shriky RC, Merrill JT. Dual antibody reactivity to beta2-glycoprotein I and protein S: increased association with thrombotic events in the antiphospholipid syndrome. Lupus 2002; 11 (04) 215-220
- 170 Ginsberg JS, Demers C, Brill-Edwards P. et al. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am J Med 1995; 98 (04) 379-383
- 171 Guermazi S, Regnault V, Gorgi Y, Ayed K, Lecompte T, Dellagi K. Further evidence for the presence of anti-protein S autoantibodies in patients with systemic lupus erythematosus. Blood Coagul Fibrinolysis 2000; 11 (05) 491-498
- 172 Song KS, Park YS, Kim HK. Prevalence of anti-protein S antibodies in patients with systemic lupus erythematosus. Arthritis Rheum 2000; 43 (03) 557-560
- 173 Wun T, Brunson A. Sickle cell disease: an inherited thrombophilia. Hematology (Am Soc Hematol Educ Program) 2016; 2016 (01) 640-647
- 174 Noubouossie D, Key NS, Ataga KI. Coagulation abnormalities of sickle cell disease: relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev 2016; 30 (04) 245-256
- 175 Henkens CM, Bom VJ, Van der Schaaf W. et al. Plasma levels of protein S, protein C, and factor X: effects of sex, hormonal state and age. Thromb Haemost 1995; 74 (05) 1271-1275
- 176 Ignjatovic V, Kenet G, Monagle P. Perinatal and Paediatric Haemostasis Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Developmental hemostasis: recommendations for laboratories reporting pediatric samples. J Thromb Haemost 2012; 10 (02) 298-300
- 177 Mulder R, Ten Kate MK, Kluin-Nelemans HC, Mulder AB. Low cut-off values increase diagnostic performance of protein S assays. Thromb Haemost 2010; 104 (03) 618-625
- 178 Nicolaes GA, Dahlbäck B. Congenital and acquired activated protein C resistance. Semin Vasc Med 2003; 3 (01) 33-46
- 179 Moore GW, Van Cott EM, Cutler JA, Mitchell MJ, Adcock DM. subcommittee on plasma coagulation inhibitors. Recommendations for clinical laboratory testing of activated protein C resistance; communication from the SSC of the ISTH. J Thromb Haemost 2019; 17 (09) 1555-1561
- 180 Bertina RM, Koeleman BP, Koster T. et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369 (6475): 64-67
- 181 Moore GW, Castoldi E, Teruya J, Morishita E, Adcock DM, Factor V. Factor V Leiden-independent activated protein C resistance: communication from the plasma coagulation inhibitors subcommittee of the International Society on Thrombosis and Haemostasis Scientific and Standardisation Committee. J Thromb Haemost 2023; 21 (01) 164-174
- 182 Sheppard DR. Activated protein C resistance: the most common risk factor for venous thromboembolism. J Am Board Fam Pract 2000; 13 (02) 111-115
- 183 Williamson D, Brown K, Luddington R, Baglin C, Baglin T, Factor V. Factor V Cambridge: a new mutation (Arg306–>Thr) associated with resistance to activated protein C. Blood 1998; 91 (04) 1140-1144
- 184 Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91 (04) 1135-1139
- 185 Cai H, Hua B, Fan L, Wang Q, Wang S, Zhao Y. A novel mutation (g2172–>c) in the factor V gene in a Chinese family with hereditary activated protein C resistance. Thromb Res 2010; 125 (06) 545-548
- 186 Gandrille S, Greengard JS, Alhenc-Gelas M. et al. Incidence of activated protein C resistance caused by the ARG 506 GLN mutation in factor V in 113 unrelated symptomatic protein C-deficient patients. The French Network on the behalf of INSERM. Blood 1995; 86 (01) 219-224
- 187 Mumford AD, McVey JH, Morse CV. et al. Factor V I359T: a novel mutation associated with thrombosis and resistance to activated protein C. Br J Haematol 2003; 123 (03) 496-501
- 188 Pezeshkpoor B, Castoldi E, Mahler A. et al. Identification and functional characterization of a novel F5 mutation (Ala512Val, FVB onn) associated with activated protein C resistance. J Thromb Haemost 2016; 14 (07) 1353-1363
- 189 Nogami K, Shinozawa K, Ogiwara K. et al. Novel FV mutation (W1920R, FVNara) associated with serious deep vein thrombosis and more potent APC resistance relative to FVLeiden. Blood 2014; 123 (15) 2420-2428
- 190 Castoldi E, Hézard N, Mourey G. et al. Severe thrombophilia in a factor V-deficient patient homozygous for the Ala2086Asp mutation (FV Besançon). J Thromb Haemost 2021; 19 (05) 1186-1199
- 191 Castoldi E, Rosing J. APC resistance: biological basis and acquired influences. J Thromb Haemost 2010; 8 (03) 445-453
- 192 Castaman G, Tosetto A, Simioni M, Ruggeri M, Madeo D, Rodeghiero F. Phenotypic APC resistance in carriers of the A20210 prothrombin mutation is associated with an increased risk of venous thrombosis. Thromb Haemost 2001; 86 (03) 804-808
- 193 Graf LL, Welsh CH, Qamar Z, Marlar RA. Activated protein C resistance assay detects thrombotic risk factors other than factor V Leiden. Am J Clin Pathol 2003; 119 (01) 52-60
- 194 Simioni P, Cagnin S, Sartorello F. et al. Partial F8 gene duplication (factor VIII Padua) associated with high factor VIII levels and familial thrombophilia. Blood 2021; 137 (17) 2383-2393
- 195 Tripodi A, Chantarangkul V, Mannucci PM. Hyperprothrombinemia may result in scquired activated protein C resistance. Blood 2000; 96 (09) 3295-3296
- 196 de Visser MC, van Hylckama Vlieg A, Tans G. et al. Determinants of the APTT- and ETP-based APC sensitivity tests. J Thromb Haemost 2005; 3 (07) 1488-1494
- 197 Sarig G, Michaeli Y, Lanir N, Brenner B, Haim N. Mechanisms for acquired activated protein C resistance in cancer patients. J Thromb Haemost 2005; 3 (03) 589-590
- 198 Negaard HF, Iversen PO, Ostenstad B, Mowinckel MC, Sandset PM. Increased acquired activated protein C resistance in unselected patients with hematological malignancies. J Thromb Haemost 2008; 6 (09) 1482-1487
- 199 Wahl D, Membre A, Perret-Guillaume C, Regnault V, Lecompte T. Mechanisms of antiphospholipid-induced thrombosis: effects on the protein C system. Curr Rheumatol Rep 2009; 11 (01) 77-81
- 200 Kadauke S, Khor B, Van Cott EM. Activated protein C resistance testing for factor V Leiden. Am J Hematol 2014; 89 (12) 1147-1150
- 201 Moore GW, Chege E, Culhane AP, Hunt BJ. Maximising the diagnostic potential of APTT-based screening assays for activated protein C resistance. Int J Lab Hematol 2015; 37 (06) 844-852
- 202 Saenz AJ, Johnson NV, Van Cott EM. Acquired activated protein C resistance caused by lupus anticoagulants. Am J Clin Pathol 2011; 136 (03) 344-349
- 203 Ragland BD, Reed CE, Eiland BM. et al. The effect of lupus anticoagulant in the second-generation assay for activated protein C resistance. Am J Clin Pathol 2003; 119 (01) 66-71
- 204 Valverde S, Trabuio E, Antico F, Manoni F, Gessoni G. Evaluation of two functional assays in detection of factor V Leiden carriers. I J La M 2009; 5: 212-216
- 205 Quehenberger P, Handler S, Mannhalter C, Kyrle PA, Speiser W. The Factor V (Leiden) test: evaluation of an assay based on dilute Russell Viper Venom Time for the detection of the Factor V Leiden mutation. Thromb Res 1999; 96 (02) 125-133
- 206 Quenhenberger P, Handler S, Mannhalter C, Pabinger-Fasching I, Speiser W. Evaluation of a highly specific functional test for the detection of factor V Leiden. Int J Clin Lab Res 2000; 30 (03) 113-117
- 207 Coen Herak D, Milos M, Zadro R. Improvement of STA:STACLOT APC-R Test for the detection of factor V Leiden by using APCR ratio. J Thromb Haemost 2005; 3 (Suppl. 01) abstract P1292
- 208 Schöni R, Quehenberger P, Wu JR, Wilmer M. Clinical evaluation of a new functional test for detection of activated protein C resistance (Pefakit APC-R Factor V Leiden) at two centers in Europe and the USA. Thromb Res 2007; 119 (01) 17-26
- 209 Amiral J, Vissac AM, Seghatchian J. Laboratory assessment of activated protein C resistance/factor V-Leiden and performance characteristics of a new quantitative assay. Transfus Apheresis Sci 2017; 56 (06) 906-913
- 210 Tripodi A, Chantarangkul V, Negri B, Mannucci PM. Standardization of the APC resistance test. Effects of normalization of results by means of pooled normal plasma. Thromb Haemost 1998; 79 (03) 564-566
- 211 Wilmer M, Stocker C, Bühler B, Conell B, Calatzis A. Improved distinction of factor V wild-type and factor V Leiden using a novel prothrombin-based activated protein C resistance assay. Am J Clin Pathol 2004; 122 (06) 836-842
- 212 Foster PA, Varma RR. Phenotypic correction of activated protein C resistance following orthotopic liver transplantation. Blood Coagul Fibrinolysis 1996; 7 (01) 65-68
- 213 Leroy-Matheron C, Duvoux C, Van Nhieu JT, Leroy K, Cherqui D, Gouault-Heilmann M. Activated protein C resistance acquired through liver transplantation and associated with recurrent venous thrombosis. J Hepatol 2003; 38 (06) 866-869
- 214 Willems M, Sterneck M, Langer F. et al. Recurrent deep-vein thrombosis based on homozygous factor V Leiden mutation acquired after liver transplantation. Liver Transpl 2003; 9 (08) 870-873
- 215 Ayala R, Martínez-López J, Cedena T. et al. Recipient and donor thrombophilia and the risk of portal venous thrombosis and hepatic artery thrombosis in liver recipients. BMC Gastroenterol 2011; 11: 130
- 216 Chiusolo P, Sica S, Salutari P. et al. Factor V Leiden and allogeneic bone marrow transplantation: chimerism as a confounding factor in genetic test interpretation. Clin Lab Haematol 1999; 21 (06) 401-402
- 217 Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfibrinogenemia. Arch Pathol Lab Med 2002; 126 (04) 499-505
- 218 Thorsen LI, Brosstad F, Solum NO, Stormorken H. Increased binding to ADP-stimulated platelets and aggregation effect of the dysfibrinogen Oslo I as compared with normal fibrinogen. Scand J Haematol 1986; 36 (02) 203-210
- 219 Shapiro SE. Diagnosis and management of dysfibrinogenemia. Clin Adv Hematol Oncol 2018; 16 (09) 602-605
- 220 Franchini M, Martinelli I, Mannucci PM. Uncertain thrombophilia markers. Thromb Haemost 2016; 115 (01) 25-30
- 221 Sachs UJ, Kirsch-Altena A, Müller J. Markers of hereditary thrombophilia with unclear significance. Hamostaseologie 2022; 42 (06) 370-380
- 222 Simioni P, Tormene D, Tognin G. et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med 2009; 361 (17) 1671-1675
- 223 Miyawaki Y, Suzuki A, Fujita J. et al. Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med 2012; 366 (25) 2390-2396
- 224 Djordjevic V, Kovac M, Miljic P. et al. A novel prothrombin mutation in two families with prominent thrombophilia–the first cases of antithrombin resistance in a Caucasian population. J Thromb Haemost 2013; 11 (10) 1936-1939
- 225 Bulato C, Radu CM, Campello E. et al. New prothrombin mutation (Arg596Trp, Prothrombin Padua 2) associated with venous thromboembolism. Arterioscler Thromb Vasc Biol 2016; 36 (05) 1022-1029
- 226 Sivasundar S, Oommen AT, Prakash O. et al. Molecular defect of 'Prothrombin Amrita': substitution of arginine by glutamine (Arg553 to Gln) near the Na(+) binding loop of prothrombin. Blood Cells Mol Dis 2013; 50 (03) 182-183
- 227 Tamura S, Suga Y, Tanamura M. et al. Optimisation of antithrombin resistance assay as a practical clinical laboratory test: development of prothrombin activator using factors Xa/Va and automation of assay. Int J Lab Hematol 2018; 40 (03) 312-319
- 228 Gosselin RC, Marlar RA. Preanalytical variables in coagulation testing: setting the stage for accurate results. Semin Thromb Hemost 2019; 45 (05) 433-448
- 229 Favaloro EJ, Gosselin RC, Pasalic L, Lippi G. Post-analytical Issues in hemostasis and thrombosis testing: an update. Methods Mol Biol 2023; 2663: 787-811
- 230 Hubbard AR. International biological standards for coagulation factors and inhibitors. Semin Thromb Hemost 2007; 33 (03) 283-289
- 231 Mackie I, Cooper P, Kitchen S. Quality assurance issues and interpretation of assays. Semin Hematol 2007; 44 (02) 114-125
- 232 Jerrard-Dunne P, Evans A, McGovern R. et al. Ethnic differences in markers of thrombophilia: implications for the investigation of ischemic stroke in multiethnic populations: the South London Ethnicity and Stroke Study. Stroke 2003; 34 (08) 1821-1826
- 233 Chen TY, Su WC, Tsao CJ. Incidence of thrombophilia detected in southern Taiwanese patients with venous thrombosis. Ann Hematol 2003; 82 (02) 114-117
- 234 Kinoshita S, Iida H, Inoue S. et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem 2005; 38 (10) 908-915
- 235 Zhu T, Ding Q, Bai X. et al. Normal ranges and genetic variants of antithrombin, protein C and protein S in the general Chinese population. Results of the Chinese Hemostasis Investigation on Natural Anticoagulants Study I Group. Haematologica 2011; 96 (07) 1033-1040
- 236 Tang L, Lu X, Yu JM. et al. PROC c.574_576del polymorphism: a common genetic risk factor for venous thrombosis in the Chinese population. J Thromb Haemost 2012; 10 (10) 2019-2026
- 237 Ormesher L, Simcox LE, Tower C, Greer IA. 'To test or not to test', the arguments for and against thrombophilia testing in obstetrics. Obstet Med 2017; 10 (02) 61-66
Address for correspondence
Publication History
Article published online:
11 May 2024
© 2024. Thieme. All rights reserved.
Thieme Medical Publishers, Inc.
333 Seventh Avenue, 18th Floor, New York, NY 10001, USA
-
References
- 1 Colucci G, Tsakiris DA. Thrombophilia screening revisited: an issue of personalized medicine. J Thromb Thrombolysis 2020; 49 (04) 618-629
- 2 Colucci G, Tsakiris DA. Thrombophilia screening: universal, selected, or neither?. Clin Appl Thromb Hemost 2017; 23 (08) 893-899
- 3 Srivastava A, Brewer AK, Mauser-Bunschoten EP. et al; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19 (01) e1-e47
- 4 Heit JA. Thrombophilia: common questions on laboratory assessment and management. Hematology (Am Soc Hematol Educ Program) 2007; 2007: 127-135
- 5 Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood 2008; 112 (01) 19-27
- 6 Zöller B, Svensson PJ, Dahlbäck B, Lind-Hallden C, Hallden C, Elf J. Genetic risk factors for venous thromboembolism. Expert Rev Hematol 2020; 13 (09) 971-981
- 7 Arachchillage DJ, Mackillop L, Chandratheva A, Motawani J, MacCallum P, Laffan M. Thrombophilia testing: a British Society for Haematology guideline. Br J Haematol 2022; 198 (03) 443-458
- 8 Mannucci PM, Franchini M. Classic thrombophilic gene variants. Thromb Haemost 2015; 114 (05) 885-889
- 9 Van Cott EM, Khor B, Zehnder JL, Factor V. Factor V Leiden. Am J Hematol 2016; 91 (01) 46-49
- 10 Rolla R, Pergolini P, Vidali M. et al. Routine coagulation tests are not useful as a screening tool for the FII G20210A polymorphism. Clin Lab 2014; 60 (10) 1725-1733
- 11 Nicolaes GA, Dahlbäck B. Activated protein C resistance (FV(Leiden)) and thrombosis: factor V mutations causing hypercoagulable states. Hematol Oncol Clin North Am 2003; 17 (01) 37-61 , vi
- 12 Kujovich JL, Factor V. Factor V Leiden thrombophilia. Genet Med 2011; 13 (01) 1-16
- 13 Middeldorp S, Nieuwlaat R, Baumann Kreuziger L. et al. American Society of Hematology 2023 guidelines for management of venous thromboembolism: thrombophilia testing. Blood Adv 2023; 7 (22) 7101-7138
- 14 Kudo M, Lee HL, Yang IA, Masel PJ. Utility of thrombophilia testing in patients with venous thrombo-embolism. J Thorac Dis 2016; 8 (12) 3697-3703
- 15 Connors JM. Thrombophilia testing and venous thrombosis. N Engl J Med 2017; 377 (12) 1177-1187
- 16 De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost 2013; 110 (04) 697-705
- 17 Stevens SM, Woller SC, Bauer KA. et al. Guidance for the evaluation and treatment of hereditary and acquired thrombophilia. J Thromb Thrombolysis 2016; 41 (01) 154-164
- 18 MacCallum P, Bowles L, Keeling D. Diagnosis and management of heritable thrombophilias. BMJ 2014; 349: g4387
- 19 Middeldorp S. Inherited thrombophilia: a double-edged sword. Hematology (Am Soc Hematol Educ Program) 2016; 2016 (01) 1-9
- 20 Franchini M. The utility of thrombophilia testing. Clin Chem Lab Med 2014; 52 (04) 495-497
- 21 Fell C, Ivanovic N, Johnson SA, Seegers WH. Differentiation of plasma antithrombin activities. Proc Soc Exp Biol Med 1954; 85 (02) 199-202
- 22 Kottke-Marchant K, Duncan A. Antithrombin deficiency: issues in laboratory diagnosis. Arch Pathol Lab Med 2002; 126 (11) 1326-1336
- 23 Rezaie AR, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost 2020; 18 (12) 3142-3153
- 24 Muszbek L, Bereczky Z, Kovács B, Komáromi I. Antithrombin deficiency and its laboratory diagnosis. Clin Chem Lab Med 2010; 48 (Suppl. 01) S67-S78
- 25 Pabinger I, Thaler J. How I treat patients with hereditary antithrombin deficiency. Blood 2019; 134 (26) 2346-2353
- 26 Law RH, Zhang Q, McGowan S. et al. An overview of the serpin superfamily. Genome Biol 2006; 7 (05) 216
- 27 Opal SM, Kessler CM, Roemisch J, Knaub S. Antithrombin, heparin, and heparan sulfate. Crit Care Med 2002; 30 (5, Suppl): S325-S331
- 28 Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci U S A 1997; 94 (26) 14683-14688
- 29 Liu J, Pedersen LC. Anticoagulant heparan sulfate: structural specificity and biosynthesis. Appl Microbiol Biotechnol 2007; 74 (02) 263-272
- 30 Swedenborg J. The mechanisms of action of alpha- and beta-isoforms of antithrombin. Blood Coagul Fibrinolysis 1998; 9 (Suppl. 03) S7-S10
- 31 Teitel JM, Rosenberg RD. Protection of factor Xa from neutralization by the heparin-antithrombin complex. J Clin Invest 1983; 71 (05) 1383-1391
- 32 Weitz JI. Heparan sulfate: antithrombotic or not?. J Clin Invest 2003; 111 (07) 952-954
- 33 Egeberg O. Thrombophilia caused by inheritable deficiency of blood antithrombin. Scand J Clin Lab Invest 1965; 17: 92
- 34 Bravo-Pérez C, de la Morena-Barrio ME, Vicente V, Corral J. Antithrombin deficiency as a still underdiagnosed thrombophilia: a primer for internists. Pol Arch Intern Med 2020; 130 (10) 868-877
- 35 Alhenc-Gelas M, Plu-Bureau G, Hugon-Rodin J, Picard V, Horellou MH. GFHT study group on Genetic Thrombophilia. Thrombotic risk according to SERPINC1 genotype in a large cohort of subjects with antithrombin inherited deficiency. Thromb Haemost 2017; 117 (06) 1040-1051
- 36 Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia 2008; 14 (06) 1229-1239
- 37 Corral J, de la Morena-Barrio ME, Vicente V. The genetics of antithrombin. Thromb Res 2018; 169: 23-29
- 38 Brown SA, Mitchell M, Cutler JA, Moore G, Smith MP, Savidge GF. Rapid genetic diagnosis in neonatal pulmonary artery thrombosis caused by homozygous antithrombin Budapest 3. Clin Appl Thromb Hemost 2000; 6 (03) 181-183
- 39 Van Cott EM, Orlando C, Moore GW, Cooper PC, Meijer P, Marlar R. Subcommittee on Plasma Coagulation Inhibitors. Recommendations for clinical laboratory testing for antithrombin deficiency; communication from the SSC of the ISTH. J Thromb Haemost 2020; 18 (01) 17-22
- 40 Cooper PC, Coath F, Daly ME, Makris M. The phenotypic and genetic assessment of antithrombin deficiency. Int J Lab Hematol 2011; 33 (03) 227-237
- 41 Kovács B, Bereczky Z, Oláh Z. et al. The superiority of anti-FXa assay over anti-FIIa assay in detecting heparin-binding site antithrombin deficiency. Am J Clin Pathol 2013; 140 (05) 675-679
- 42 Moser KA, Smock KJ. Direct oral anticoagulant (DOAC) interference in hemostasis assays. Hematology (Am Soc Hematol Educ Program) 2021; 2021 (01) 129-133
- 43 Smogorzewska A, Brandt JT, Chandler WL. et al. Effect of fondaparinux on coagulation assays: results of College of American Pathologists proficiency testing. Arch Pathol Lab Med 2006; 130 (11) 1605-1611
- 44 Van Blerk M, Bailleul E, Chatelain B. et al. Influence of dabigatran and rivaroxaban on routine coagulation assays. A nationwide Belgian survey. Thromb Haemost 2015; 113 (01) 154-164
- 45 Favresse J, Lardinois B, Sabor L. et al. Evaluation of the DOAC-Stop® procedure to overcome the effect of DOACs on several thrombophilia screening tests. TH Open 2018; 2 (02) e202-e209
- 46 Antovic J, Söderström J, Karlman B, Blombäck M. Evaluation of a new immunoturbidimetric test (Liatest antithrombin III) for determination of antithrombin antigen. Clin Lab Haematol 2001; 23 (05) 313-316
- 47 Heit JA. Thrombophilia: clinical and laboratory assessment and management. In: Kitchens CS, Alving BM, Kessler CM. eds. Consultative Hemostasis and Thrombosis. 2nd ed.. Philadelphia: Saunders; 2007: 213-244
- 48 Kovács B, Bereczky Z, Selmeczi A. et al. Progressive chromogenic anti-factor Xa assay and its use in the classification of antithrombin deficiencies. Clin Chem Lab Med 2014; 52 (12) 1797-1806
- 49 Chowdhury V, Mille B, Olds RJ. et al. Antithrombins Southport (Leu 99 to Val) and Vienna (Gln 118 to Pro): two novel antithrombin variants with abnormal heparin binding. Br J Haematol 1995; 89 (03) 602-609
- 50 Moore GW, de Jager N, Cutler JA. Development of a novel, rapid assay for detection of heparin-binding defect antithrombin deficiencies: the heparin-antithrombin binding (HAB) ratio. Thromb Res 2015; 135 (01) 161-166
- 51 Corral J, Hernandez-Espinosa D, Soria JM. et al. Antithrombin Cambridge II (A384S): an underestimated genetic risk factor for venous thrombosis. Blood 2007; 109 (10) 4258-4263
- 52 Perry DJ, Daly ME, Tait RC. et al. Antithrombin Cambridge II (Ala384Ser): clinical, functional and haplotype analysis of 18 families. Thromb Haemost 1998; 79 (02) 249-253
- 53 Olson ST, Stephens AW, Hirs CH, Bock PE, Björk I. Kinetic characterization of the proteinase binding defect in a reactive site variant of the serpin, antithrombin. Role of the P1′ residue in transition-state stabilization of antithrombin-proteinase complex formation. J Biol Chem 1995; 270 (17) 9717-9724
- 54 Ungerstedt JS, Schulman S, Egberg N, Antovic J, Blombäck N. Discrepancy between antithrombin activity methods revealed in antithrombin Stockholm: do factor Xa-based methods overestimate antithrombin activity in some patients?. Blood 2002; 99 (06) 2271-2272
- 55 Lane DA, Lowe GD, Flynn A, Thompson E, Ireland H, Erdjument H. Antithrombin III Glasgow: a variant with increased heparin affinity and reduced ability to inactivate thrombin, associated with familial thrombosis. Br J Haematol 1987; 66 (04) 523-527
- 56 Beauchamp NJ, Pike RN, Daly M. et al. Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood 1998; 92 (08) 2696-2706
- 57 Bravo-Pérez C, de la Morena-Barrio ME, de la Morena-Barrio B. et al. Molecular and clinical characterization of transient antithrombin deficiency: a new concept in congenital thrombophilia. Am J Hematol 2022; 97 (02) 216-225
- 58 Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn–>Asp). J Clin Invest 1994; 94 (06) 2265-2274
- 59 Picard V, Bauters A, Khairy M. et al. Conformational Asn187Asp/Lys antithrombin variants and thrombosis. Clinical and biological features in 13 new heterozygotes. Thromb Haemost 2005; 93 (01) 57-62
- 60 Navarro-Fernández J, de la Morena-Barrio ME, Padilla J. et al. Antithrombin Dublin (p.Val30Glu): a relatively common variant with moderate thrombosis risk of causing transient antithrombin deficiency. Thromb Haemost 2016; 116 (01) 146-154
- 61 Weronska A, De la Morena-Barrio B, Goldman-Mazur S. et al. Functional, biochemical, molecular and clinical characterization of antithrombin c.1157T>C (p.Ile386Thr), a recurrent Polish variant with a founder effect. Haematologica 2023; 108 (10) 2803-2807
- 62 de la Morena-Barrio ME, Suchon P, Jacobsen EM. et al. Two SERPINC1 variants affecting N-glycosylation of Asn224 cause severe thrombophilia not detected by functional assays. Blood 2022; 140 (02) 140-151
- 63 Goldenberg NA, Manco-Johnson MJ. Protein C deficiency. Haemophilia 2008; 14 (06) 1214-1221
- 64 Stenflo J. A new vitamin K-dependent protein. Purification from bovine plasma and preliminary characterization. J Biol Chem 1976; 251 (02) 355-363
- 65 Dahlbäck B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol 2005; 25 (07) 1311-1320
- 66 Thiyagarajan M, Cheng T, Zlokovic BV. Endothelial cell protein C receptor: role beyond endothelium?. Circ Res 2007; 100 (02) 155-157
- 67 Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018; 132 (02) 148-158
- 68 Kottke-Marchant K, Comp P. Laboratory issues in diagnosing abnormalities of protein C, thrombomodulin, and endothelial cell protein C receptor. Arch Pathol Lab Med 2002; 126 (11) 1337-1348
- 69 Nicolaes GA, Dahlbäck B. Factor V and thrombotic disease: description of a Janus-faced protein. Arterioscler Thromb Vasc Biol 2002; 22 (04) 530-538
- 70 Dinarvand P, Moser KA, Protein C. Deficiency. Arch Pathol Lab Med 2019; 143 (10) 1281-1285
- 71 Bahou WF. Thrombin's faces revealed: cellular effects include induction of neoangiogenesis. J Thromb Haemost 2003; 1 (10) 2078-2080
- 72 MacMahon A. The realm of Janus: Doorways in the Roman World. In: Carr G, Swift E, Weekes J. eds. TRAC 2002: Proceedings of the Twelfth Annual Theoretical Roman Archaeology Conference, Canterbury 2002. Oxford: Oxbow Books; 2003: 58-73
- 73 Suzuki K. The multi-functional serpin, protein C inhibitor: beyond thrombosis and hemostasis. J Thromb Haemost 2008; 6 (12) 2017-2026
- 74 Marlar RA, Gausman JN. Laboratory testing issues for protein C, protein S, and antithrombin. Int J Lab Hematol 2014; 36 (03) 289-295
- 75 Minford A, Brandão LR, Othman M. et al. Diagnosis and management of severe congenital protein C deficiency (SCPCD): communication from the SSC of the ISTH. J Thromb Haemost 2022; 20 (07) 1735-1743
- 76 Tripodi A, Franchi F, Krachmalnicoff A, Mannucci PM. Asymptomatic homozygous protein C deficiency. Acta Haematol 1990; 83 (03) 152-155
- 77 Shakoor MT, Ayub S, Ayub Z. Warfarin induced skin necrosis: a diagnostic challenge. MOJ Clin Med Case Rep 2017; 6: 21-22
- 78 Khor B, Van Cott EM. Laboratory tests for protein C deficiency. Am J Hematol 2010; 85 (06) 440-442
- 79 Cooper PC, Pavlova A, Moore GW, Hickey KP, Marlar RA. Recommendations for clinical laboratory testing for protein C deficiency, for the subcommittee on plasma coagulation inhibitors of the ISTH. J Thromb Haemost 2020; 18 (02) 271-277
- 80 Moore GW. Snake venoms in diagnostic hemostasis and thrombosis. Semin Thromb Hemost 2022; 48 (02) 145-160
- 81 Cooper PC, Hill M, Maclean RM. The phenotypic and genetic assessment of protein C deficiency. Int J Lab Hematol 2012; 34 (04) 336-346
- 82 Simioni P, Lazzaro A, Zanardi S, Girolami A. Spurious protein C deficiency due to antiphospholipid antibodies. Am J Hematol 1991; 36 (04) 299-301
- 83 Favre R, Zia-Chahabi S, Talb Y, de Gunzburg N, Flaujac C. Direct oral anticoagulant neutralization by activated charcoal DOAC-remove for thrombophilia screening. Blood Coagul Fibrinolysis 2021; 32 (05) 356-358
- 84 Ruinemans-Koerts J, Peterse-Stienissen I, Verbruggen B. Non-parallelism in the one-stage coagulation factor assay is a phenomenon of lupus anticoagulants and not of individual factor inhibitors. Thromb Haemost 2010; 104 (05) 1080-1082
- 85 Mackie IJ, Gallimore M, Machin SJ. Contact factor proteases and the complexes formed with alpha 2-macroglobulin can interfere in protein C assays by cleaving amidolytic substrates. Blood Coagul Fibrinolysis 1992; 3 (05) 589-595
- 86 Han P, Fung KP, Rahdakrishnan U. Lack of agreement of chromogenic and clotting assays for factor X and protein C in warfarinised plasma. Thromb Haemost 1991; 65 (04) 360-363
- 87 Bovill EG, Tomczak JA, Grant B. et al. Protein CVermont: symptomatic type II protein C deficiency associated with two GLA domain mutations. Blood 1992; 79 (06) 1456-1465
- 88 Gaussem P, Gandrille S, Duchemin J. et al. Influence of six mutations of the protein C gene on the Gla domain conformation and calcium affinity. Thromb Haemost 1994; 71 (06) 748-754
- 89 Seidel H, Haracska B, Naumann J, Westhofen P, Hass MS, Kruppenbacher JP. Laboratory limitations of excluding hereditary protein C deficiency by chromogenic assay: discrepancies of phenotype and genotype. Clin Appl Thromb Hemost 2020; 26: 1076029620912028
- 90 Cooper PC, Siddiq S, Morse C, Nightingale J, Mumford AD. Marked discrepancy between coagulometric protein C activity assays with the pro-thrombotic protein C Asn2Ile substitution. Int J Lab Hematol 2011; 33 (05) 451-456
- 91 Noguchi K, Nakazono E, Tsuda T. et al. Plasma phenotypes of protein S Lys196Glu and protein C Lys193del variants prevalent among young Japanese women. Blood Coagul Fibrinolysis 2019; 30 (08) 393-400
- 92 Efthymiou M, Arachchillage DRJ, Lane PJ. et al. Antibodies against TFPI and protein C are associated with a severe thrombotic phenotype in patients with and without antiphospholipid syndrome. Thromb Res 2018; 170: 60-68
- 93 MacCallum PK, Cooper JA, Martin J, Howarth DJ, Meade TW, Miller GJ. Associations of protein C and protein S with serum lipid concentrations. Br J Haematol 1998; 102 (02) 609-615
- 94 Cushman M, Psaty BM, Meilahn EN, Dobs AS, Kuller LH. Post-menopausal hormone therapy and concentrations of protein C and antithrombin in elderly women. Br J Haematol 2001; 114 (01) 162-168
- 95 Said JM, Ignjatovic V, Monagle PT, Walker SP, Higgins JR, Brennecke SP. Altered reference ranges for protein C and protein S during early pregnancy: implications for the diagnosis of protein C and protein S deficiency during pregnancy. Thromb Haemost 2010; 103 (05) 984-988
- 96 Monagle P, Barnes C, Ignjatovic V. et al. Developmental haemostasis. Impact for clinical haemostasis laboratories. Thromb Haemost 2006; 95 (02) 362-372
- 97 Toulon P. Developmental hemostasis: laboratory and clinical implications. Int J Lab Hematol 2016; 38 (Suppl. 01) 66-77
- 98 Appel IM, Grimminck B, Geerts J, Stigter R, Cnossen MH, Beishuizen A. Age dependency of coagulation parameters during childhood and puberty. J Thromb Haemost 2012; 10 (11) 2254-2263
- 99 Andrew M, Paes B, Milner R. et al. Development of the human coagulation system in the full-term infant. Blood 1987; 70 (01) 165-172
- 100 Andrew M, Vegh P, Johnston M, Bowker J, Ofosu F, Mitchell L. Maturation of the hemostatic system during childhood. Blood 1992; 80 (08) 1998-2005
- 101 Toulon P, Berruyer M, Brionne-François M. et al. Age dependency for coagulation parameters in paediatric populations. Results of a multicentre study aimed at defining the age-specific reference ranges. Thromb Haemost 2016; 116 (01) 9-16
- 102 Attard C, van der Straaten T, Karlaftis V, Monagle P, Ignjatovic V. Developmental hemostasis: age-specific differences in the levels of hemostatic proteins. J Thromb Haemost 2013; 11 (10) 1850-1854
- 103 Conlan MG, Folsom AR, Finch A, Davis CE, Sorlie P, Wu KK. Correlation of plasma protein C levels with cardiovascular risk factors in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) study. Thromb Haemost 1993; 70 (05) 762-767
- 104 Tait RC, Walker ID, Islam SI. et al. Protein C activity in healthy volunteers–influence of age, sex, smoking and oral contraceptives. Thromb Haemost 1993; 70 (02) 281-285
- 105 Lowe GD, Rumley A, Woodward M. et al. Epidemiology of coagulation factors, inhibitors and activation markers: the Third Glasgow MONICA Survey. I. Illustrative reference ranges by age, sex and hormone use. Br J Haematol 1997; 97 (04) 775-784
- 106 Franchi F, Biguzzi E, Martinelli I. et al. Normal reference ranges of antithrombin, protein C and protein S: effect of sex, age and hormonal status. Thromb Res 2013; 132 (02) e152-e157
- 107 Rodeghiero F, Tosetto A. The VITA Project: population-based distributions of protein C, antithrombin III, heparin-cofactor II and plasminogen–relationship with physiological variables and establishment of reference ranges. Thromb Haemost 1996; 76 (02) 226-233
- 108 Allaart CF, Poort SR, Rosendaal FR, Reitsma PH, Bertina RM, Briët E. Increased risk of venous thrombosis in carriers of hereditary protein C deficiency defect. Lancet 1993; 341 (8838): 134-138
- 109 Di Scipio RG, Hermodson MA, Yates SG, Davie EW. A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry 1977; 16 (04) 698-706
- 110 Ahnström J, Andersson HM, Canis K. et al. Activated protein C cofactor function of protein S: a novel role for a γ-carboxyglutamic acid residue. Blood 2011; 117 (24) 6685-6693
- 111 Hackeng TM, Maurissen LF, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost 2009; 7 (Suppl. 01) 165-168
- 112 Gierula M, Ahnström J. Anticoagulant protein S-New insights on interactions and functions. J Thromb Haemost 2020; 18 (11) 2801-2811
- 113 Dahlbäck B, Guo LJ, Livaja-Koshiar R, Tran S. Factor V-short and protein S as synergistic tissue factor pathway inhibitor (TFPIα) cofactors. Res Pract Thromb Haemost 2017; 2 (01) 114-124
- 114 Gierula M, Noakes VM, Salles-Crawley II, Crawley JTB, Ahnström J. The TFPIα C-terminal tail is essential for TFPIα-FV-short-protein S complex formation and synergistic enhancement of TFPIα. J Thromb Haemost 2023; 21 (12) 3568-3580
- 115 Dahlbäck B. The tale of protein S and C4b-binding protein, a story of affection. Thromb Haemost 2007; 98 (01) 90-96
- 116 Maurissen LF, Thomassen MC, Nicolaes GA. et al. Re-evaluation of the role of the protein S-C4b binding protein complex in activated protein C-catalyzed factor Va-inactivation. Blood 2008; 111 (06) 3034-3041
- 117 ten Kate MK, van der Meer J. Protein S deficiency: a clinical perspective. Haemophilia 2008; 14 (06) 1222-1228
- 118 Marlar RA, Gausman JN, Tsuda H, Rollins-Raval MA, Brinkman HJM. Recommendations for clinical laboratory testing for protein S deficiency: communication from the SSC committee plasma coagulation inhibitors of the ISTH. J Thromb Haemost 2021; 19 (01) 68-74
- 119 Zöller B, García de Frutos P, Dahlbäck B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood 1995; 85 (12) 3524-3531
- 120 Marlar RA, Gausman JN. Protein S abnormalities: a diagnostic nightmare. Am J Hematol 2011; 86 (05) 418-421
- 121 Pintao MC, Ribeiro DD, Bezemer ID. et al. Protein S levels and the risk of venous thrombosis: results from the MEGA case-control study. Blood 2013; 122 (18) 3210-3219
- 122 Anderson DR, Brill-Edwards P, Walker I. Warfarin-induced skin necrosis in 2 patients with protein S deficiency: successful reinstatement of warfarin therapy. Haemostasis 1992; 22 (03) 124-128
- 123 Tai CY, Ierardi R, Alexander JB. A case of warfarin skin necrosis despite enoxaparin anticoagulation in a patient with protein S deficiency. Ann Vasc Surg 2004; 18 (02) 237-242
- 124 Fraga R, Diniz LM, Lucas EA, Emerich PS. Warfarin-induced skin necrosis in a patient with protein S deficiency. An Bras Dermatol 2018; 93 (04) 612-613
- 125 Tsuda T, Jin X, Tsuda H. et al. New quantitative total protein S-assay system for diagnosing protein S type II deficiency: clinical application of the screening system for protein S type II deficiency. Blood Coagul Fibrinolysis 2012; 23 (01) 56-63
- 126 Goodwin AJ, Rosendaal FR, Kottke-Marchant K, Bovill EG. A review of the technical, diagnostic, and epidemiologic considerations for protein S assays. Arch Pathol Lab Med 2002; 126 (11) 1349-1366
- 127 Smock KJ, Plumhoff EA, Meijer P. et al. Protein S testing in patients with protein S deficiency, factor V Leiden, and rivaroxaban by North American Specialized Coagulation Laboratories. Thromb Haemost 2016; 116 (01) 50-57
- 128 Lawrie AS, Lloyd ME, Mohamed F, Irons S, Hughes GR, Savidge GF. Assay of protein S in systemic lupus erythematosus. Blood Coagul Fibrinolysis 1995; 6 (04) 322-324
- 129 Rossi E, Gatti L, Guarneri D, Finotto E, Lombardi A, Preda L. Functional protein S in women with lupus anticoagulant inhibitor. Thromb Res 1992; 65 (02) 253-262
- 130 Tomás JF, Alberca I, Tabernero MD, Cordero M, Del Pino-Montes J, Vicente V. Natural anticoagulant proteins and antiphospholipid antibodies in systemic lupus erythematosus. J Rheumatol 1998; 25 (01) 57-62
- 131 Seriolo B, Cutolo M, Accardo S. Association between acquired free protein S deficiency, anticardiolipin antibodies, and thrombotic events in rheumatoid arthritis. J Rheumatol 1998; 25 (11) 2281-2282
- 132 Parke AL, Weinstein RE, Bona RD, Maier DB, Walker FJ. The thrombotic diathesis associated with the presence of phospholipid antibodies may be due to low levels of free protein S. Am J Med 1992; 93 (01) 49-56
- 133 Van Cott EM, Ledford-Kraemer M, Meijer P, Nichols WL, Johnson SM, Peerschke EI. NASCOLA Proficiency Testing Committee. Protein S assays: an analysis of North American Specialized Coagulation Laboratory Association proficiency testing. Am J Clin Pathol 2005; 123 (05) 778-785
- 134 Jennings I, Kitchen S, Woods TA, Preston FE. Multilaboratory testing in thrombophilia through the United Kingdom National External Quality Assessment Scheme (Blood Coagulation) Quality Assurance Program. Semin Thromb Hemost 2005; 31 (01) 66-72
- 135 Jennings I, Kitchen S, Cooper P, Makris M, Preston FE. Sensitivity of functional protein S assays to protein S deficiency: a comparative study of three commercial kits. J Thromb Haemost 2003; 1 (05) 1112-1114
- 136 Marlar RA, Potts RM, Welsh CW. Accuracy of diagnosis of protein S deficiency by protein S activity and antigen assays. J Clin Ligand 2005; 28: 130-135
- 137 Rodger MA, Carrier M, Gervais M, Rock G. Normal functional protein S activity does not exclude protein S deficiency. Pathophysiol Haemost Thromb 2003; 33 (04) 202-205
- 138 Siriez R, Dogné JM, Gosselin R, Laloy J, Mullier F, Douxfils J. Comprehensive review of the impact of direct oral anticoagulants on thrombophilia diagnostic tests: practical recommendations for the laboratory. Int J Lab Hematol 2021; 43 (01) 7-20
- 139 Bovill EG, Landesman MM, Busch SA, Fregeau GR, Mann KG, Tracy RP. Studies on the measurement of protein S in plasma. Clin Chem 1991; 37 (10 Pt 1): 1708-1714
- 140 Hubbard AR, Wong MY. A collaborative study on the measurement of protein S antigen in plasma. Thromb Haemost 1992; 68 (02) 115-118
- 141 Persson KE, Hillarp A, Dahlbäck B. Analytical considerations for free protein S assays in protein S deficiency. Thromb Haemost 2001; 86 (05) 1144-1147
- 142 Duebgen S, Kauke T, Marschall C. et al. Genotype and laboratory and clinical phenotypes of protein s deficiency. Am J Clin Pathol 2012; 137 (02) 178-184
- 143 Brinkman HJM, Ahnström J, Castoldi E, Dahlbäck B, Marlar RA. Pleiotropic anticoagulant functions of protein S, consequences for the clinical laboratory. Communication from the SSC of the ISTH. J Thromb Haemost 2021; 19 (01) 281-286
- 144 Bertina RM, Ploos van Amstel HK, van Wijngaarden A. et al. Heerlen polymorphism of protein S, an immunologic polymorphism due to dimorphism of residue 460. Blood 1990; 76 (03) 538-548
- 145 Duchemin J, Gandrille S, Borgel D. et al. The Ser 460 to Pro substitution of the protein S alpha (PROS1) gene is a frequent mutation associated with free protein S (type IIa) deficiency. Blood 1995; 86 (09) 3436-3443
- 146 Varvenne M, Kochhan L, Trummer A, Eisert R, Birschmann I. Clinical consequences of compound heterozygosity for protein S mutation Heerlen and p.Cys252Gly protein S mutation. Thromb Res 2011; 128 (05) 498-500
- 147 Wypasek E, Potaczek DP, Alhenc-Gelas M, Undas A. Heerlen polymorphism associated with type III protein S deficiency and factor V Leiden mutation in a Polish patient with deep vein thrombosis. Blood Coagul Fibrinolysis 2014; 25 (01) 84-85
- 148 Suchon P, Germain M, Delluc A. et al. Protein S Heerlen mutation heterozygosity is associated with venous thrombosis risk. Sci Rep 2017; 7: 45507
- 149 Espinosa-Parrilla Y, Navarro G, Morell M, Abella E, Estivill X, Sala N. Homozygosity for the protein S Heerlen allele is associated with type I PS deficiency in a thrombophilic pedigree with multiple risk factors. Thromb Haemost 2000; 83 (01) 102-106
- 150 Alhenc-Gelas M, Canonico M, Morange PE, Emmerich J. Geht Genetic Thrombophilia Group. Protein S inherited qualitative deficiency: novel mutations and phenotypic influence. J Thromb Haemost 2010; 8 (12) 2718-2726
- 151 Denis CV, Roberts SJ, Hackeng TM, Lenting PJ. In vivo clearance of human protein S in a mouse model: influence of C4b-binding protein and the Heerlen polymorphism. Arterioscler Thromb Vasc Biol 2005; 25 (10) 2209-2215
- 152 Heeb MJ, Koenen RR, Fernández JA, Hackeng TM. Direct anticoagulant activity of protein S-C4b binding protein complex in Heerlen heterozygotes and normals. J Thromb Haemost 2004; 2 (10) 1766-1773
- 153 Hayashi T, Nishioka J, Suzuki K. Molecular mechanism of the dysfunction of protein S(Tokushima) (Lys155–>Glu) for the regulation of the blood coagulation system. Biochim Biophys Acta 1995; 1272 (03) 159-167
- 154 D'Angelo A, Viganò D'Angelo S. Protein S deficiency. Haematologica 2008; 93 (04) 498-501
- 155 Vigano-D'Angelo S, D'Angelo A, Kaufman Jr CE, Sholer C, Esmon CT, Comp PC. Protein S deficiency occurs in the nephrotic syndrome. Ann Intern Med 1987; 107 (01) 42-47
- 156 Ceriello A, Giugliano D, Quatraro A, Marchi E, Barbanti M, Lefebvre P. Possible role for increased C4b-binding-protein level in acquired protein S deficiency in type I diabetes. Diabetes 1990; 39 (04) 447-449
- 157 Addai-Mensah O, Annani-Akollor ME, Nsafoah FO. et al. Effect of poor glycaemic control on plasma levels and activity of protein C, protein S, and antithrombin III in type 2 diabetes mellitus. PLoS One 2019; 14 (09) e0223171
- 158 Fernández JA, Estellés A, Gilabert J, España F, Aznar J. Functional and immunologic protein S in normal pregnant women and in full-term newborns. Thromb Haemost 1989; 61 (03) 474-478
- 159 Fu M, Liu J, Xing J. et al. Reference intervals for coagulation parameters in non-pregnant and pregnant women. Sci Rep 2022; 12 (01) 1519
- 160 Szecsi PB, Jørgensen M, Klajnbard A, Andersen MR, Colov NP, Stender S. Haemostatic reference intervals in pregnancy. Thromb Haemost 2010; 103 (04) 718-727
- 161 Oruç S, Saruç M, Koyuncu FM, Ozdemir E. Changes in the plasma activities of protein C and protein S during pregnancy. Aust N Z J Obstet Gynaecol 2000; 40 (04) 448-450
- 162 Stahl CP, Wideman CS, Spira TJ, Haff EC, Hixon GJ, Evatt BL. Protein S deficiency in men with long-term human immunodeficiency virus infection. Blood 1993; 81 (07) 1801-1807
- 163 Lafeuillade A, Sorice M, Griggi T, Pellegrino P, Geoffroy M, Profizi N. Role of autoimmunity in protein S deficiency during HIV-1 infection. Infection 1994; 22 (03) 201-203
- 164 D'Angelo A, Della Valle P, Crippa L, Pattarini E, Grimaldi LM, Viganò D'Angelo S. Brief report: autoimmune protein S deficiency in a boy with severe thromboembolic disease. N Engl J Med 1993; 328 (24) 1753-1757
- 165 Regnault V, Boehlen F, Ozsahin H. et al. Anti-protein S antibodies following a varicella infection: detection, characterization and influence on thrombin generation. J Thromb Haemost 2005; 3 (06) 1243-1249
- 166 Deitcher SR, Erban JK, Limentani SA. Acquired free protein S deficiency associated with multiple myeloma: a case report. Am J Hematol 1996; 51 (04) 319-323
- 167 Guermazi S, Hamza M, Dellagi K. Protein S deficiency and antibodies to protein S in patients with Behçet's disease. Thromb Res 1997; 86 (03) 197-204
- 168 Crowther MA, Johnston M, Weitz J, Ginsberg JS. Free protein S deficiency may be found in patients with antiphospholipid antibodies who do not have systemic lupus erythematosus. Thromb Haemost 1996; 76 (05) 689-691
- 169 Erkan D, Zhang HW, Shriky RC, Merrill JT. Dual antibody reactivity to beta2-glycoprotein I and protein S: increased association with thrombotic events in the antiphospholipid syndrome. Lupus 2002; 11 (04) 215-220
- 170 Ginsberg JS, Demers C, Brill-Edwards P. et al. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am J Med 1995; 98 (04) 379-383
- 171 Guermazi S, Regnault V, Gorgi Y, Ayed K, Lecompte T, Dellagi K. Further evidence for the presence of anti-protein S autoantibodies in patients with systemic lupus erythematosus. Blood Coagul Fibrinolysis 2000; 11 (05) 491-498
- 172 Song KS, Park YS, Kim HK. Prevalence of anti-protein S antibodies in patients with systemic lupus erythematosus. Arthritis Rheum 2000; 43 (03) 557-560
- 173 Wun T, Brunson A. Sickle cell disease: an inherited thrombophilia. Hematology (Am Soc Hematol Educ Program) 2016; 2016 (01) 640-647
- 174 Noubouossie D, Key NS, Ataga KI. Coagulation abnormalities of sickle cell disease: relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev 2016; 30 (04) 245-256
- 175 Henkens CM, Bom VJ, Van der Schaaf W. et al. Plasma levels of protein S, protein C, and factor X: effects of sex, hormonal state and age. Thromb Haemost 1995; 74 (05) 1271-1275
- 176 Ignjatovic V, Kenet G, Monagle P. Perinatal and Paediatric Haemostasis Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Developmental hemostasis: recommendations for laboratories reporting pediatric samples. J Thromb Haemost 2012; 10 (02) 298-300
- 177 Mulder R, Ten Kate MK, Kluin-Nelemans HC, Mulder AB. Low cut-off values increase diagnostic performance of protein S assays. Thromb Haemost 2010; 104 (03) 618-625
- 178 Nicolaes GA, Dahlbäck B. Congenital and acquired activated protein C resistance. Semin Vasc Med 2003; 3 (01) 33-46
- 179 Moore GW, Van Cott EM, Cutler JA, Mitchell MJ, Adcock DM. subcommittee on plasma coagulation inhibitors. Recommendations for clinical laboratory testing of activated protein C resistance; communication from the SSC of the ISTH. J Thromb Haemost 2019; 17 (09) 1555-1561
- 180 Bertina RM, Koeleman BP, Koster T. et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994; 369 (6475): 64-67
- 181 Moore GW, Castoldi E, Teruya J, Morishita E, Adcock DM, Factor V. Factor V Leiden-independent activated protein C resistance: communication from the plasma coagulation inhibitors subcommittee of the International Society on Thrombosis and Haemostasis Scientific and Standardisation Committee. J Thromb Haemost 2023; 21 (01) 164-174
- 182 Sheppard DR. Activated protein C resistance: the most common risk factor for venous thromboembolism. J Am Board Fam Pract 2000; 13 (02) 111-115
- 183 Williamson D, Brown K, Luddington R, Baglin C, Baglin T, Factor V. Factor V Cambridge: a new mutation (Arg306–>Thr) associated with resistance to activated protein C. Blood 1998; 91 (04) 1140-1144
- 184 Chan WP, Lee CK, Kwong YL, Lam CK, Liang R. A novel mutation of Arg306 of factor V gene in Hong Kong Chinese. Blood 1998; 91 (04) 1135-1139
- 185 Cai H, Hua B, Fan L, Wang Q, Wang S, Zhao Y. A novel mutation (g2172–>c) in the factor V gene in a Chinese family with hereditary activated protein C resistance. Thromb Res 2010; 125 (06) 545-548
- 186 Gandrille S, Greengard JS, Alhenc-Gelas M. et al. Incidence of activated protein C resistance caused by the ARG 506 GLN mutation in factor V in 113 unrelated symptomatic protein C-deficient patients. The French Network on the behalf of INSERM. Blood 1995; 86 (01) 219-224
- 187 Mumford AD, McVey JH, Morse CV. et al. Factor V I359T: a novel mutation associated with thrombosis and resistance to activated protein C. Br J Haematol 2003; 123 (03) 496-501
- 188 Pezeshkpoor B, Castoldi E, Mahler A. et al. Identification and functional characterization of a novel F5 mutation (Ala512Val, FVB onn) associated with activated protein C resistance. J Thromb Haemost 2016; 14 (07) 1353-1363
- 189 Nogami K, Shinozawa K, Ogiwara K. et al. Novel FV mutation (W1920R, FVNara) associated with serious deep vein thrombosis and more potent APC resistance relative to FVLeiden. Blood 2014; 123 (15) 2420-2428
- 190 Castoldi E, Hézard N, Mourey G. et al. Severe thrombophilia in a factor V-deficient patient homozygous for the Ala2086Asp mutation (FV Besançon). J Thromb Haemost 2021; 19 (05) 1186-1199
- 191 Castoldi E, Rosing J. APC resistance: biological basis and acquired influences. J Thromb Haemost 2010; 8 (03) 445-453
- 192 Castaman G, Tosetto A, Simioni M, Ruggeri M, Madeo D, Rodeghiero F. Phenotypic APC resistance in carriers of the A20210 prothrombin mutation is associated with an increased risk of venous thrombosis. Thromb Haemost 2001; 86 (03) 804-808
- 193 Graf LL, Welsh CH, Qamar Z, Marlar RA. Activated protein C resistance assay detects thrombotic risk factors other than factor V Leiden. Am J Clin Pathol 2003; 119 (01) 52-60
- 194 Simioni P, Cagnin S, Sartorello F. et al. Partial F8 gene duplication (factor VIII Padua) associated with high factor VIII levels and familial thrombophilia. Blood 2021; 137 (17) 2383-2393
- 195 Tripodi A, Chantarangkul V, Mannucci PM. Hyperprothrombinemia may result in scquired activated protein C resistance. Blood 2000; 96 (09) 3295-3296
- 196 de Visser MC, van Hylckama Vlieg A, Tans G. et al. Determinants of the APTT- and ETP-based APC sensitivity tests. J Thromb Haemost 2005; 3 (07) 1488-1494
- 197 Sarig G, Michaeli Y, Lanir N, Brenner B, Haim N. Mechanisms for acquired activated protein C resistance in cancer patients. J Thromb Haemost 2005; 3 (03) 589-590
- 198 Negaard HF, Iversen PO, Ostenstad B, Mowinckel MC, Sandset PM. Increased acquired activated protein C resistance in unselected patients with hematological malignancies. J Thromb Haemost 2008; 6 (09) 1482-1487
- 199 Wahl D, Membre A, Perret-Guillaume C, Regnault V, Lecompte T. Mechanisms of antiphospholipid-induced thrombosis: effects on the protein C system. Curr Rheumatol Rep 2009; 11 (01) 77-81
- 200 Kadauke S, Khor B, Van Cott EM. Activated protein C resistance testing for factor V Leiden. Am J Hematol 2014; 89 (12) 1147-1150
- 201 Moore GW, Chege E, Culhane AP, Hunt BJ. Maximising the diagnostic potential of APTT-based screening assays for activated protein C resistance. Int J Lab Hematol 2015; 37 (06) 844-852
- 202 Saenz AJ, Johnson NV, Van Cott EM. Acquired activated protein C resistance caused by lupus anticoagulants. Am J Clin Pathol 2011; 136 (03) 344-349
- 203 Ragland BD, Reed CE, Eiland BM. et al. The effect of lupus anticoagulant in the second-generation assay for activated protein C resistance. Am J Clin Pathol 2003; 119 (01) 66-71
- 204 Valverde S, Trabuio E, Antico F, Manoni F, Gessoni G. Evaluation of two functional assays in detection of factor V Leiden carriers. I J La M 2009; 5: 212-216
- 205 Quehenberger P, Handler S, Mannhalter C, Kyrle PA, Speiser W. The Factor V (Leiden) test: evaluation of an assay based on dilute Russell Viper Venom Time for the detection of the Factor V Leiden mutation. Thromb Res 1999; 96 (02) 125-133
- 206 Quenhenberger P, Handler S, Mannhalter C, Pabinger-Fasching I, Speiser W. Evaluation of a highly specific functional test for the detection of factor V Leiden. Int J Clin Lab Res 2000; 30 (03) 113-117
- 207 Coen Herak D, Milos M, Zadro R. Improvement of STA:STACLOT APC-R Test for the detection of factor V Leiden by using APCR ratio. J Thromb Haemost 2005; 3 (Suppl. 01) abstract P1292
- 208 Schöni R, Quehenberger P, Wu JR, Wilmer M. Clinical evaluation of a new functional test for detection of activated protein C resistance (Pefakit APC-R Factor V Leiden) at two centers in Europe and the USA. Thromb Res 2007; 119 (01) 17-26
- 209 Amiral J, Vissac AM, Seghatchian J. Laboratory assessment of activated protein C resistance/factor V-Leiden and performance characteristics of a new quantitative assay. Transfus Apheresis Sci 2017; 56 (06) 906-913
- 210 Tripodi A, Chantarangkul V, Negri B, Mannucci PM. Standardization of the APC resistance test. Effects of normalization of results by means of pooled normal plasma. Thromb Haemost 1998; 79 (03) 564-566
- 211 Wilmer M, Stocker C, Bühler B, Conell B, Calatzis A. Improved distinction of factor V wild-type and factor V Leiden using a novel prothrombin-based activated protein C resistance assay. Am J Clin Pathol 2004; 122 (06) 836-842
- 212 Foster PA, Varma RR. Phenotypic correction of activated protein C resistance following orthotopic liver transplantation. Blood Coagul Fibrinolysis 1996; 7 (01) 65-68
- 213 Leroy-Matheron C, Duvoux C, Van Nhieu JT, Leroy K, Cherqui D, Gouault-Heilmann M. Activated protein C resistance acquired through liver transplantation and associated with recurrent venous thrombosis. J Hepatol 2003; 38 (06) 866-869
- 214 Willems M, Sterneck M, Langer F. et al. Recurrent deep-vein thrombosis based on homozygous factor V Leiden mutation acquired after liver transplantation. Liver Transpl 2003; 9 (08) 870-873
- 215 Ayala R, Martínez-López J, Cedena T. et al. Recipient and donor thrombophilia and the risk of portal venous thrombosis and hepatic artery thrombosis in liver recipients. BMC Gastroenterol 2011; 11: 130
- 216 Chiusolo P, Sica S, Salutari P. et al. Factor V Leiden and allogeneic bone marrow transplantation: chimerism as a confounding factor in genetic test interpretation. Clin Lab Haematol 1999; 21 (06) 401-402
- 217 Cunningham MT, Brandt JT, Laposata M, Olson JD. Laboratory diagnosis of dysfibrinogenemia. Arch Pathol Lab Med 2002; 126 (04) 499-505
- 218 Thorsen LI, Brosstad F, Solum NO, Stormorken H. Increased binding to ADP-stimulated platelets and aggregation effect of the dysfibrinogen Oslo I as compared with normal fibrinogen. Scand J Haematol 1986; 36 (02) 203-210
- 219 Shapiro SE. Diagnosis and management of dysfibrinogenemia. Clin Adv Hematol Oncol 2018; 16 (09) 602-605
- 220 Franchini M, Martinelli I, Mannucci PM. Uncertain thrombophilia markers. Thromb Haemost 2016; 115 (01) 25-30
- 221 Sachs UJ, Kirsch-Altena A, Müller J. Markers of hereditary thrombophilia with unclear significance. Hamostaseologie 2022; 42 (06) 370-380
- 222 Simioni P, Tormene D, Tognin G. et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med 2009; 361 (17) 1671-1675
- 223 Miyawaki Y, Suzuki A, Fujita J. et al. Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med 2012; 366 (25) 2390-2396
- 224 Djordjevic V, Kovac M, Miljic P. et al. A novel prothrombin mutation in two families with prominent thrombophilia–the first cases of antithrombin resistance in a Caucasian population. J Thromb Haemost 2013; 11 (10) 1936-1939
- 225 Bulato C, Radu CM, Campello E. et al. New prothrombin mutation (Arg596Trp, Prothrombin Padua 2) associated with venous thromboembolism. Arterioscler Thromb Vasc Biol 2016; 36 (05) 1022-1029
- 226 Sivasundar S, Oommen AT, Prakash O. et al. Molecular defect of 'Prothrombin Amrita': substitution of arginine by glutamine (Arg553 to Gln) near the Na(+) binding loop of prothrombin. Blood Cells Mol Dis 2013; 50 (03) 182-183
- 227 Tamura S, Suga Y, Tanamura M. et al. Optimisation of antithrombin resistance assay as a practical clinical laboratory test: development of prothrombin activator using factors Xa/Va and automation of assay. Int J Lab Hematol 2018; 40 (03) 312-319
- 228 Gosselin RC, Marlar RA. Preanalytical variables in coagulation testing: setting the stage for accurate results. Semin Thromb Hemost 2019; 45 (05) 433-448
- 229 Favaloro EJ, Gosselin RC, Pasalic L, Lippi G. Post-analytical Issues in hemostasis and thrombosis testing: an update. Methods Mol Biol 2023; 2663: 787-811
- 230 Hubbard AR. International biological standards for coagulation factors and inhibitors. Semin Thromb Hemost 2007; 33 (03) 283-289
- 231 Mackie I, Cooper P, Kitchen S. Quality assurance issues and interpretation of assays. Semin Hematol 2007; 44 (02) 114-125
- 232 Jerrard-Dunne P, Evans A, McGovern R. et al. Ethnic differences in markers of thrombophilia: implications for the investigation of ischemic stroke in multiethnic populations: the South London Ethnicity and Stroke Study. Stroke 2003; 34 (08) 1821-1826
- 233 Chen TY, Su WC, Tsao CJ. Incidence of thrombophilia detected in southern Taiwanese patients with venous thrombosis. Ann Hematol 2003; 82 (02) 114-117
- 234 Kinoshita S, Iida H, Inoue S. et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem 2005; 38 (10) 908-915
- 235 Zhu T, Ding Q, Bai X. et al. Normal ranges and genetic variants of antithrombin, protein C and protein S in the general Chinese population. Results of the Chinese Hemostasis Investigation on Natural Anticoagulants Study I Group. Haematologica 2011; 96 (07) 1033-1040
- 236 Tang L, Lu X, Yu JM. et al. PROC c.574_576del polymorphism: a common genetic risk factor for venous thrombosis in the Chinese population. J Thromb Haemost 2012; 10 (10) 2019-2026
- 237 Ormesher L, Simcox LE, Tower C, Greer IA. 'To test or not to test', the arguments for and against thrombophilia testing in obstetrics. Obstet Med 2017; 10 (02) 61-66