
Subscribe to RSS
DOI: 10.1055/s-0042-108619
Ichthyosiformer Morphenwandel einer erythrodermen Mycosis fungoides unter extrakorporaler Photopherese-Therapie – klinisch relevant?
Erythrodermic Mycosis Fungoides Presenting with Ichthyosiform Eruptions Under Extracorporal PhotopheresisKorrespondenzadresse
Publication History
Publication Date:
13 October 2016 (online)

Zusammenfassung
Die Mycosis fungoides (MF) tritt klassischerweise in drei Stadien (Patch-, Plaque- und Tumorstadium) auf und ist ein den Dermatologen äußerst geläufiges Krankheitsbild. Zahlreiche morphologische Varianten der MF sind bekannt, von denen drei als separate Varianten in der WHO/EORTC-Klassifikation 2005 aufgeführt werden (follikulotrope MF, pagetoide Retikulose und die sog. „granulomatous slack skin“). Erythroderme Varianten der MF sind von einem Sézary-Syndrom abzugrenzen, für welches definierte Diagnosekriterien existieren. Selten erst wurde über einen ichthyosiformen Subtyp der MF berichtet. Wir stellen den Verlauf einer erythrodermen MF vor, welche unter einer extrakorporalen Photopheresetherapie einen ichthyosiformen Morphenwandel zeigte, diskutieren sowohl klinische als auch histologische Merkmale und stellen pathogenetische Hypothesen zur Diskussion.
#
Abstract
Mycosis fungoides (MF) typically manifests in three classical clinical stages (patch, plaque and tumor form) and its clinical presentation is well known for dermatologists and dermatopathologists. A great variety of morphologic variants are known and three distinct entities have been named in the WHO/EORTC classification system in 2005: folliculotropic MF, pagetoid reticulosis and granulomatous slack skin. Erythrodermic forms are known and have to be differentiated from a Sézary syndrome. Reports of an ichthyosiform MF are rare. We present the case of an erythrodermic variant of MF with development of ichthyosiform eruptions under extracorporal photopheresis. Clinical and dermatopathological aspects as well as pathogenetic hypothesis are discussed.
#
Fallvorstellung
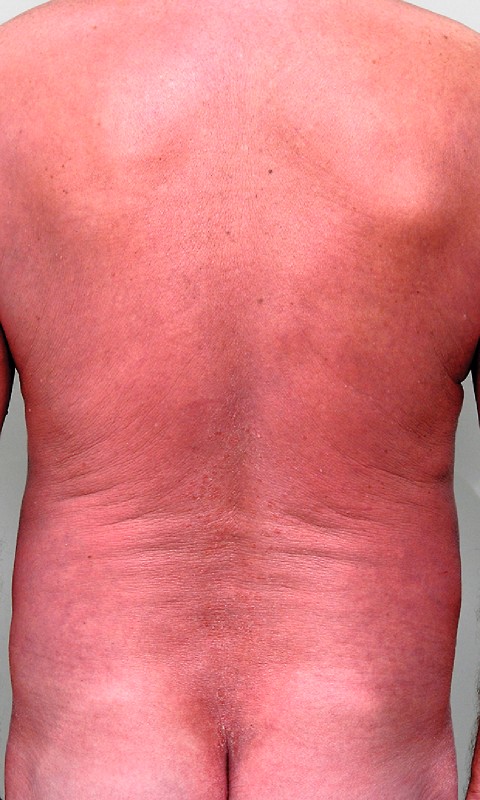
Es wird über einen 65-jährigen Patienten berichtet, der sich initial im Januar 2015 mit seit ca. sechs Monaten bestehenden stark juckenden Hautveränderungen in unserer Klinik vorstellte. Die dermatologische Eigen- und Familienanamnese war leer. Zu sehen war eine Erythrodermie mit generalisierter Lichenifikation, an den Plantae leicht ausgeprägte Hyperkeratosen ([Abb. 1]). Rechts axillär und inguinal beidseits konnten Lymphknotenpakete palpiert werden, welche sonografisch als reaktive bzw. dermatopathische Lymphknoten beurteilt wurden. Ambulant war der Patient topisch mit diversen kortisonhaltigen handelsüblichen Präparaten behandelt worden, ohne eine Besserung des Hautbefundes zu erzielen. In einer Hautbiopsie von der Flanke zeigte sich eine teils orthokeratotisch, teils parakeratotisch verhornende, regelrecht geschichtete Epidermis mit leichter epidermaler Akanthose. Subepidermal fand sich ein lichenoides Entzündungsinfiltrat aus recht monomorphen Lymphozyten. Fokal infiltrierten einzelne Lymphozyten in die Epidermis im Sinne eines positiven Epidermotropismus unter Ausbildung von zahlreichen Pautrierʼschen Mikroabszessen ([Abb. 4 a]). In Zusammenschau mit den klinischen Befunden einer (Sub-)Erythrodermie, Lymphadenopathie, bei jedoch fehlendem Nachweis von Sézary-Zellen (CD4+/CD26−Subpopulation von T-Zellen mit zerebriformen Zellkernen [1]) im peripheren Blut diagnostizierten wir eine erythroderme Mycosis fungoides (MF).
Primär wurde neben einer topischen Therapie mit steroidhaltigen Externa eine extrakorporale Photopherese (ECP) eingeleitet. Diese erfolgte zunächst in 14-tägigen, später 4-wöchigen Intervallen. Unter dieser Therapie konnte eine zügige Abblassung der Haut unter Hinterlassung flächiger, postinflammatorischer Hyperpigmentierungen erreicht werden. Nach insgesamt 6 Zyklen der ECP-Behandlung zeigte sich bei dem Patienten ein zunehmender morphologischer Wandel des Hautbefundes mit flächigen, bräunlichen, nicht schuppenden, ichthyosiformen, polygonalen, festhaftenden Keratosen ohne Juckreiz unter Aussparung der großen Körperbeugen und der Körperfalten (sog. „deck-chair-Sign“) in Analogie zum Papuloerythroderma Ofuji ([Abb. 2 a – c]) [2]. Die durchgeführte Hautbiopsie vom Abdomen zeigte Cluster aus großzelligen, CD30+-atypischen, lymphoiden Zellen im dermalen Infiltrat ([Abb. 3a – c]). In [Abb. 4] werden Detailaufnahmen gezeigt, welche insbesondere das fokal recht schmale Stratum granulosum in der Biopsie aus der ichthyotischen Läsion zeigen ([Abb. 4 b]). In der Zusammenschau der erhobenen klinischen und histologischen Befunde war nun von einer ichthyosiformen Mycosis fungoides mit großzelliger Transformation auszugehen.



#
Diskussion
Die World Health Organisation (WHO) und European Organisation for Research and Treatment of Cancer (EORTC) haben 2005 eine neue Klassifikation der Mycosis fungoides (MF) veröffentlicht [3]. Hierin werden neben der klassischen Mycosis fungoides mit Patch-, Plaque- und Tumorstadium drei distinkte Varianten genannt und separat gelistet:
-
die follikulotrope Variante mit charakteristischen follikulotropen T-Zell-Infiltraten unter Aussparungen der Epidermis und Prädilektion des Kopf- und Nackenbereiches
-
die pagetoide Retikulose mit Patches und Plaques und intraepidermaler Proliferation von neoplastischen T-Zellen (Morbus Woringer-Kolopp)
-
die „granulomatous slack skin“-Variante, als sehr seltene Form der MF, welche in charakteristischer Weise in den Hautfalten auftritt, zu „sackartiger“ Dermatochalasis führt und histologisch ganz distinkte granulomatöse Infiltrate mehrkerniger T-Zellen und eine Phagozytose elastischer Bindegewebsfasern aufweist.
Von der MF abzugrenzen ist das SS, welches auch in der WHO-Klassifikation als eigenständige Entität gelistet ist und durch Erythrodermie mit schwerem therapierefraktärem Juckreiz, Lymphadenopathie und leukämischer Aussaat von sog. Sézary-Zellen gekennzeichnet ist. Weitere definierte kutane T-Zell-Lymphome umfassen die adulte T-Zell-Leukämie, CD30-positive lymphoproliferative Erkrankungen sowie pannikulitische Lymphome [3].
Die International Society for Cutaneous Lymphomas (ISCL) und die EORTC haben 2011 ein Staging-System für die MF und das Sézary-Syndrom (SS) auf Basis der TNM-Klassifikation aufgestellt und hieraus ein standardisiertes diagnostisches Einteilungssystem entwickelt, woraus sich zukünftige Therapieansätze ableiten, auf die hier nicht detailliert eingegangen werden soll [4].
Im vorliegenden Fall möchten wir über einen klinisch imposanten Wandel der Morphe einer MF berichten, mit Elementen der ausgeprägten Ichthyosis vulgaris und des Papuloerythroderma Ofuji („deck chair sign“) ([Abb. 2 a – c]), welche eine relativ „konventionelle“, d. h. nicht spezifische Histologie der MF aufweist. Die ichthyosiforme MF (iMF) als solche ist bekannt und tritt in 1,8 – 3,5 % der Patienten mit einer MF auf [5] [6] [7] [8] [9]. Sie kann nochmals in 3 Subvarianten eingeteilt werden: 1) ichthyosiforme Eruptionen als solitäre Manifestation der Erkrankung, 2) ichthyosiforme Eruption in Verbindung mit zusätzlichen atypischen Anzeichen einer MF und 3) ichthyosiforme Eruption in Kombination mit der klassischen Variante der MF [9].
Klinisch manifestiert sich die iMF mit hyperpigmentierten, xerotisch-schuppenden sowie flachen ichthyosiformen Plaques. Kazakov und Kollegen beschreiben zusätzliche begleitende Komedo-artige Läsionen und/oder follikuläre, keratotische Papeln, die bei unserem Patienten nicht auftraten [7] [10]. Histologisch weist die iMF die typischen Kennzeichen einer konventionellen MF auf. Immunphänotypisch liegt meist ein Helfer-Phänotyp mit CD2+/CD3+/CD4+- und CD45RO+-Expression bei fehlender Expression von CD7, CD20 und CD30 vor [5] [6]. In ca. 5 % einer iMF können auch prädominant CD8-positive zytotoxische T-Lymphozyten und CD19+/CD20+/CD22+-B-Lymphozyten im Infiltrat vorhanden sein [6] [10] [11]. Immunhistochemisch konnten wir bei unserem Patienten CD30+-atypische, pleomorphe, lymphoide T-Zellen nachweisen. Morizane und Kollegen beschreiben, dass die Ausbildung von CD30+-atypischen, pleomorphen, lymphoiden, T-Zellen mit einer erworbenen Ichthyose im Rahmen einer MF assoziiert sein kann und dieser Phänotyp ein Indiz für einen rasch progredienten Verlauf der Erkrankung mit schlechterer Prognose ist [8]. Des Weiteren wurde kein aggressiverer Verlauf im Falle einer iMF verglichen mit der konventionellen MF beschrieben [6]. Bislang wurden nur ca. 20 Fälle von iMF beschrieben [5] [10]. Es handelt sich also um eine sehr selten auftretende klinische Spielart der MF, welche im klinischen Alltag sicherlich beeindruckend ist, jedoch nicht a priori mit einem aggressiveren Verlauf assoziiert ist. Besondere Aufmerksamkeit verdient in unserem Fall auch das ausgeprägte „deck-chair-sign“ (DCS), welches im Zusammenhang mit der ichthyosiformen Eruption aufgetreten ist und zuvor im Rahmen der Erythrodermie nicht gesehen wurde ([Abb. 2 a]). Unter dem DCS versteht man die Aussparung von Körperfalten durch Hautläsionen. Die Pathogenese des DCS, d. h. die Frage, wie Druck in den Körperfalten vor Infiltration durch ein entzündliches oder neoplastisches Infiltrat schützt, ist derzeit nicht beantwortet. Das DCS tritt auch im Rahmen von Arzneiexanthemen auf, ist jedoch auch beim Lupus erythematosus, Kontaktdermatitiden, dem Erysipel und der Akanthosis nigricans beobachtet worden [12]. Berichte über das gemeinsame Auftreten eines DCS im Zusammenhang mit kutanen Lymphomen und im Speziellen im Zusammenhang mit ichthyosiformen Hautveränderungen sind selten [13].
Im Gegensatz zu den genetisch verursachten Formen der Ichthyose beginnt eine nicht-kongenitale, erworbene Ichthyose naturgemäß meist im Erwachsenenalter und ist assoziiert mit Malignomen, insbesondere Morbus Hodgkin, Autoimmun- und Infektionserkrankungen, Endokrinopathien, Niereninsuffizienz (z. B. IgA-Nephropathien), kann aber auch im Rahmen einer Mangelernährung und Medikamenten-assoziiert auftreten [11]. Die nicht-kongenitalen und daher erworbenen Ichthyosen zählen zu den sog. Pseudoichthyosen und können somit als eine paraneoplastische Manifestation auch einer MF aufgefasst werden [10]. Die detaillierte Pathogenese der erworbenen Ichthyosen ist bis heute ungeklärt. Es wird eine erhöhte Expression des epidermal growth factor (EGF)-Rezeptors als auch des transforming growth factor α (TGF)-Rezeptors an läsionalen Keratinozyten beschrieben [11].
In Analogie zu angeborenen Ichthyoseformen und zur atopischen Dermatitis wurde für die iMF eine deutlich verminderte Expression von Filaggrin gezeigt, welches ein Schlüsselprotein der epidermalen Differenzierung darstellt und eine ganz zentrale Rolle in der Gewährleistung einer intakten epidermalen Barrierefunktion spielt [8]. Pathogenetisch bleibt – wenngleich spekulativ – zu diskutieren, ob bei den Patienten, welche eine iMF im Verlauf entwickeln, eine Prädisposition im Sinne einer subklinisch gestörten Filaggrinexpression vorliegt, welche in Analogie zur atopischen Dermatitis einen spezifischen klinischen „ichthyotischen“ Phänotyp der Erkrankung begünstigt. Interessant ist insbesondere die Tatsache, dass sowohl kutane T-Zell-Lymphome als auch die atopische Dermatitis durch eine alterierte Filaggrinexpression (im Falle der atopischen Dermatitis auch Loricrin-Expression) eine gestörte epidermale Barrierefunktion entwickeln, welche für eine gesteigerte lokale Infektionsanfälligkeit prädisponiert (zusätzlich zu einer gestörten Induktion antimikrobieller Peptide [AMPs]) [14]. Wenngleich die extrakorporale Photopheresetherapie allein, bis auf gelegentliche katheterassoziierte Infektionen, keine klinisch relevant gesteigerte Infektionsanfälligkeit der Patienten bedingt, so sind doch Fälle beschrieben, in denen die Kombination aus ECP und medikamentöser Immunsuppression zu vital bedrohlichen Infektionen (bes. durch Krytokokken und Pilze) geführt hat [3] [15]. Dieser Fakt sollte vor dem Hintergrund des bei kutanen T-Zell-Lymphomen ohnehin gegebenen T-Zell-Defekts und der gestörten Barrierefunktion im Rahmen von kutanen T-Zell-Lymphomen im Allgemeinen und bei ichthyosiformen Erkrankungen wie der iMF im Speziellen berücksichtigt und die Patienten diesbezüglich engmaschig beobachtet werden. Wir applizieren daher bei unserem Patienten konsequent täglich Emollentien mit antiseptischem Zusatz, z. B. Triclosan 1 %.
#
Fazit
-
Die iMF stellt eine sehr seltene Variante der MF mit imposanten klinischen und klassischen histologischen Befunden dar.
-
Prognose und Verlauf der iMF scheinen nicht von denen der klassischen MF abzuweichen.
-
Die iMF scheint von einer alterierten Expression epidermaler Barriereproteine begleitet zu sein, welche zu einer verminderten Infektionsabwehr beitragen kann. Dies begründet eine engmaschige Infektionskontrolle der Patienten, v. a. im Falle einer kombinierten Therapie mit immunsuppressiven Substanzen und Methoden sowie topischen Emollentien mit Antiseptikazusatz.
-
Das Auftreten einer erythrodermen Mycosis fungoides mit Elementen des Papuloerythroderma Ofuji unter extrakorporaler Photopheresetherapie aus einem Sézary-artigen Bild kann hier erstmals dokumentiert werden.
#
#
Interessenkonflikt
Die Autoren geben an, dass kein Interessenkonflikt besteht.
-
Literatur
- 1 Fierro MT et al. Expression pattern of chemokine receptors and chemokine release in inflammatory erythroderma and Sezary syndrome. Dermatology 2006; 213: 284-292
- 2 Aste N et al. Ofuji papuloerythroderma. J Eur Acad Dermatol Venereol 2000; 14: 55-57
- 3 Willemze R et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105: 3768-3785
- 4 Olsen EA et al. Sezary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol 2011; 64: 352-404
- 5 Bianchi L et al. Ichthyosiform mycosis fungoides: a neoplastic acquired ichthyosis. Acta Derm Venereol 2007; 87: 82-83
- 6 Hodak E et al. Ichthyosiform mycosis fungoides: an atypical variant of cutaneous T-cell lymphoma. J Am Acad Dermatol 2004; 50: 368-374
- 7 Kazakov DV, Burg G, Kempf W. Clinicopathological spectrum of mycosis fungoides. J Eur Acad Dermatol Venereol 2004; 18: 397-415
- 8 Morizane S et al. Ichthyosiform eruptions in association with primary cutaneous T-cell lymphomas. Br J Dermatol 2009; 161: 115-120
- 9 Nam KH et al. Mycosis fungoides as an ichthyosiform eruption. Ann Dermatol 2009; 21: 182-184
- 10 Marzano AV et al. Ichthyosiform mycosis fungoides. Dermatology 2002; 204: 124-129
- 11 Sato M et al. Ichthyosiform mycosis fungoides: report of a case associated with IgA nephropathy. Dermatology 2005; 210: 324-328
- 12 Bettoli V et al. Ofuji papulo-erythroderma: a reappraisal of the deck-chair sign. Dermatology 2004; 209: 1-4
- 13 Adam ND. Simultaneous “deck-chair” sign and acquired ichthyosis: A novel presentation of adult T-cell leukemia/lymphoma. Journal of the American Academy of Dermatology 2007; 56: AB142
- 14 Suga H et al. Skin barrier dysfunction and low antimicrobial peptide expression in cutaneous T-cell lymphoma. Clin Cancer Res 2014; 20: 4339-4348
- 15 Frieden TR et al. Cutaneous cryptococcosis in a patient with cutaneous T cell lymphoma receiving therapy with photopheresis and methotrexate. Clin Infect Dis 1993; 17: 776-778
Korrespondenzadresse
-
Literatur
- 1 Fierro MT et al. Expression pattern of chemokine receptors and chemokine release in inflammatory erythroderma and Sezary syndrome. Dermatology 2006; 213: 284-292
- 2 Aste N et al. Ofuji papuloerythroderma. J Eur Acad Dermatol Venereol 2000; 14: 55-57
- 3 Willemze R et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105: 3768-3785
- 4 Olsen EA et al. Sezary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol 2011; 64: 352-404
- 5 Bianchi L et al. Ichthyosiform mycosis fungoides: a neoplastic acquired ichthyosis. Acta Derm Venereol 2007; 87: 82-83
- 6 Hodak E et al. Ichthyosiform mycosis fungoides: an atypical variant of cutaneous T-cell lymphoma. J Am Acad Dermatol 2004; 50: 368-374
- 7 Kazakov DV, Burg G, Kempf W. Clinicopathological spectrum of mycosis fungoides. J Eur Acad Dermatol Venereol 2004; 18: 397-415
- 8 Morizane S et al. Ichthyosiform eruptions in association with primary cutaneous T-cell lymphomas. Br J Dermatol 2009; 161: 115-120
- 9 Nam KH et al. Mycosis fungoides as an ichthyosiform eruption. Ann Dermatol 2009; 21: 182-184
- 10 Marzano AV et al. Ichthyosiform mycosis fungoides. Dermatology 2002; 204: 124-129
- 11 Sato M et al. Ichthyosiform mycosis fungoides: report of a case associated with IgA nephropathy. Dermatology 2005; 210: 324-328
- 12 Bettoli V et al. Ofuji papulo-erythroderma: a reappraisal of the deck-chair sign. Dermatology 2004; 209: 1-4
- 13 Adam ND. Simultaneous “deck-chair” sign and acquired ichthyosis: A novel presentation of adult T-cell leukemia/lymphoma. Journal of the American Academy of Dermatology 2007; 56: AB142
- 14 Suga H et al. Skin barrier dysfunction and low antimicrobial peptide expression in cutaneous T-cell lymphoma. Clin Cancer Res 2014; 20: 4339-4348
- 15 Frieden TR et al. Cutaneous cryptococcosis in a patient with cutaneous T cell lymphoma receiving therapy with photopheresis and methotrexate. Clin Infect Dis 1993; 17: 776-778