
Subscribe to RSS
DOI: 10.1055/s-0041-110199
Maligne Lymphome der Haut – Diagnostik und Therapie primär kutaner B-Zell-Lymphome in der Praxis
Malignant Lymphomas of the Skin – Diagnosis and Therapy of Primary Cutaneous B-Cell Lymphomas
- Zusammenfassung
- Abstract
- Lernziele
- Einleitung
- Primär kutanes Marginalzonenlymphom
- Primär kutanes follikuläres Lymphom (Keimzentrumslymphom)
- Primär kutanes diffuses großzelliges B-Zell-Lymphom des Unterschenkels, „leg type“
- Therapie der primär kutanen B-Zell-Lymphome
- Ausblick
- Literatur
Zusammenfassung
Primär kutane B-Zell-Lymphome (primary cutaneous B-cell lymphomas: PCBCL) stellen extranodale Non-Hodgkin-Lymphome reifer B-Lymphozyten dar, die die Haut als Zielorgan haben und dort proliferieren. Sie zeigen eine große Bandbreite klinischer und histologischer Erscheinungsformen. Ca. 22 % der kutanen Lymphome (cutaneous lymphomas: CL) entfallen auf kutane B-Zell-Lymphome (cutaneous B-cell lymphomas: CBCL), 73 % auf kutane T-Zell-Lymphome (cutaneous T-cell lymphomas: CTCL) und < 10 % werden seltenen Formen von CL zugeordnet. Die Inzidenz der CL ist weiterhin zunehmend und wird basierend auf den Daten aus den USA auf eine Neuerkrankung pro Jahr und 100 000 Einwohner geschätzt.
Die Diagnose eines PCBCL erfolgt in der Regel anhand klinischer und histologischer bzw. immunhistologischer Untersuchungen. Nicht immer jedoch erlauben diese Untersuchungen eine zweifelsfreie Diagnose. Vor allem frühe Stadien eines PCBCL ähneln sowohl klinisch als auch histologisch oft benignen entzündlichen Hauterkrankungen. Aufgrund der Heterogenität der PCBCL mit unterschiedlicher biologischer Potenz, die von einem indolenten Verlauf bis hin zu einem fatalen Verlauf reicht, ist eine eindeutige Diagnosestellung unabdingbar. Hier spielt insbesondere die Dermatohistologie, in Kombination mit der Immunhistologie und Molekularbiologie, eine entscheidende Rolle.
#
Abstract
Primary cutaneous B-cell lymphomas (PCBCL) represent extranodal non-Hodgkin’s lymphomas of mature B lymphocytes with the skin as the target organ and the site to proliferate. They show a wide range of clinical and histological features. Approximately 22 % of cutaneous lymphomas (CL) are made up of cutaneous B-cell lymphomas (CBCL) and 73 % of cutaneous T-cell lymphomas (CTCL). The remaining less than 10 % are classified as rare subtypes of CL. The incidence of CL remains increasingly and is estimated at one incidence per year and 100 000 inhabitants. The diagnosis of PCBCL is usually based on clinical, histological and immunohistochemical investigations. But the findings do not always allow an unambiguous diagnosis. Especially early stages of a PCBCL often resemble benign inflammatory skin diseases, both clinically and histologically. Due to the heterogeneity of PCBCL including different biological potencies, ranging from an indolent to a fulminant and fatal course, a distinct diagnosis is essential. Therefore, the correlation of dermatohistology, in combination with immunohistochemistry and molecular biology, plays a crucial role.
#
Lernziele
Dieser Artikel soll praxisrelevant über die häufigsten PCBCL in der Dermatologie informieren und einen Einblick in die häufigsten Krankheitsbilder, die Prognose und die Behandlungsmöglichkeiten geben.
In diesem Übersichtsartikel werden die 3 häufigsten Entitäten der PCBCL vorgestellt, das primär kutane follikuläre Lymphom (PCFCL), das primär kutane Marginalzonenlymphom (PCMZL) und das diffus großzellige B-Zell-Lymphom vom „leg-type“ (PCBLT).
#
Einleitung
Die Klassifikation der B-Zell-Lymphome wurde 2005 gemeinsam von der WHO und EORTC herausgegeben und 2008 aktualisiert [1] [2]. Diese Klassifikation ist gegenwärtig maßgeblich.
Bei den PCBCL lassen sich 2 Gruppen differenzieren. Zu den sogenannten indolenten kutanen B-Zell-Lymphom-Varianten zählen das PCFCL und das PCMZL. Bei beiden Erkrankungen ist die Lebenserwartung in aller Regel durch die Erkrankung nicht beeinträchtigt, da eine Ausbreitung der Erkrankung auf andere Organe extrem selten vorkommt. Das PCBLT hingegen zählt zu den aggressiven Lymphomen, es wird häufiger die Ausdehnung der Erkrankung auf Lymphknoten und Organe festgestellt. Die Varianten der PCBCL können oft schon klinisch anhand des Verteilungsmusters und des feingeweblichen Musters voneinander unterschieden werden. Die histologisch charakteristischen Muster, z. B. die Ausbildung sogenannter Keimzentren, hat auch zu der Benennung der einzelnen Erkrankungen beigetragen ([Tab. 1]). Zur exakten Diagnose werden heute in der Regel weitere Ergebnisse von Spezialfärbungen („Immunhistochemie“) und molekularbiologischen Untersuchungen herangezogen. Neben der üblichen Ausbreitungsdiagnostik (Ultraschalluntersuchungen von Bauch und Lymphknoten, Röntgenuntersuchung des Brustkorbs, ggf. Computertomografie) ist zusätzlich bei aggressiven Formen eine Knochenmarksbiopsie angezeigt, um auszuschließen, dass die Lymphomzellen in der Haut von einem systemischen B-Zell-Lymphom oder einer B-Zell-Leukämie abstammen.
PCBCL stellen eine heterogene Gruppe von Entitäten dar, die sich hinsichtlich der Klinik, der Histologie, des Immunphänotyps und des Genotyps unterscheiden. Die Prognose von PCBCL und PCMZL ist, bis auf wenige Ausnahmen, exzellent. Das PCBLT hingegen zählt zu den aggressiven Lymphomen mit schlechter Prognose.
#
Primär kutanes Marginalzonenlymphom
Klinisch präsentiert sich das PCMZL üblicherweise mit multifokalen, meist flachen, kleinen (< 3 cm) rötlich/violetten Papeln und Knoten ohne epidermale Beteiligung am Körperstamm und den oberen Extremitäten ([Abb. 1]) [3]. Die Tumore zeigen ein langsames Wachstum und ulzerieren in der Regel nicht. Im Gegensatz zum PCFCL ist die Kopf-/Hals-Region nur selten betroffen. Erythematöse Effloreszenzen können in der Frühform auftreten. Die Patienten klagen mitunter über Juckreiz. In der Regel werden weder die Lymphknoten noch die Organe befallen. In der Mehrzahl der Fälle tritt das PCMZL im Bereich des 40. Lebensjahr auf (median 39 Jahre).

Histologisch zeigen sich unter einer regelhaften Epidermis fleckförmige bis knotige oder diffuse Infiltrate im Bereich der gesamten Dermis, die bis an die Subkutisgrenze heranreichen und den oberen Anteil des Pannikulus miteinbeziehen können. Die neoplastische Population setzt sich typischerweise zusammen aus klein- bis mittelgroßen Zellen mit irregulären zentrozytenähnlichen Kernen und mäßig weitem Zytoplasmasaum (Marginalzonenzellen) sowie aus einigen lymphoplasmazytoiden Zellen und Plasmazellen ([Abb. 2] und [Abb. 3]). Der Anteil reaktiver Zellen am Infiltrat ist häufig hoch, wohingegen der Tumorzellanteil variabel und, die Diagnose erschwerend, sehr gering sein kann. Charakteristisch ist eine Expansion der Tumorzellen in die Umgebung reaktiver B-Follikel mit reaktiven Keimzentren (peri- oder interfollikuläres Wachstum). In fortgeschrittenen Stadien können die Follikelmäntel zunehmend von neoplastischen Zellen verdrängt und die Keimzentren von Tumorzellen durchsetzt werden.


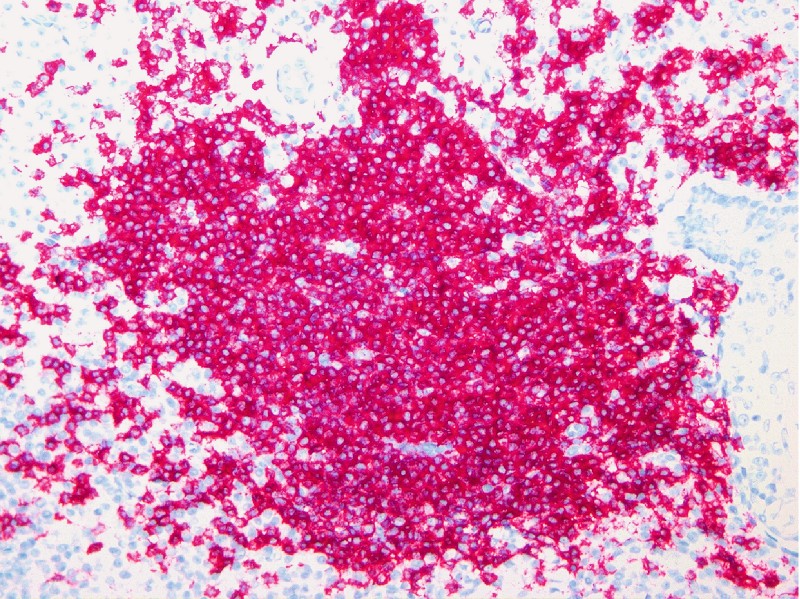
Immunhistologisch zeigen sich die Marginalzonenzellen positiv für CD20, CD79a und Bcl-2 sowie negativ für CD5, CD10 und Bcl-6 ([Abb. 4] und [Abb. 5]). CD10- und Bcl-6-Negativität kann in manchen Fällen hilfreich für die Abgrenzung des PCMZL vom PCFCL sein. Insbesondere bei plasmazellulärer Differenzierung lässt sich häufig eine monotypische intrazytoplasmatische Expression der Immunglobulin-Leichtkette nachweisen (sog. Leichtkettenrestriktion; [Abb. 6] und [Abb. 7]). Fälle mit hoher Anzahl monotypischer Plasmazellen und lymphoplasmazytoiden Zellen mit intranukleären Einschlüssen (Dutcher bodies), die früher als Immunozytom klassifiziert wurden, werden in der aktuellen WHO-Klassifikation als PCMZL bezeichnet. In fortgeschrittenen Stadien lassen sich die Reste der zerstörten Sekundärfollikel schließlich nur noch in Form aufgesplitterter Maschenwerke follikulärer dendritischer Zellen mittels Färbung für CD21 immunhistologisch identifizieren. Eine klonale Umlagerung des Immunglobulin-Schwerketten-Gens lässt sich mithilfe der PCR in mehr als 70 % der Fälle nachweisen [4].



Prognose: Trotz der häufig auftretenden kutanen Rezidive haben die Patienten meist eine exzellente Prognose (5-Jahres-Überlebensrate [JÜR] 98 %), da es in der Regel nicht zu einer systemischen Manifestation kommt. In einigen Fällen wurden Spontanremissionen beschrieben [5].
#
Primär kutanes follikuläres Lymphom (Keimzentrumslymphom)
Das PCFCL ist das häufigste B-Zell-Lymphom der Haut. Im Jahre 1951 berichtete Crosti von Patienten, die sich mit erythematösen Plaques und Knötchen in seiner Ambulanz vorgestellt hatten. Wegen der häufigen Lokalisation an Kopf, Hals, Nacken und oberem Stammbereich ([Abb. 8]) wurde der Name „follicle centre of the head and trunk“ vorgeschlagen. Aus diesem Grund wurden früher als Synonyme das Retikulohistiozytom des Rückens bzw. Crosti’s Lymphom benutzt [6]. Das Retikulohistiozytom des Rückens wurde später als primäres kutanes Keimzentrumslymphom klassifiziert: „primary cutaneous follicle center lymphoma“ (PCFCL). Die erythematösen Plaques und Knötchen treten häufig solitär auf und zeigen meist einen langsamen progredienten Verlauf. Im Verlauf zeigen sich knotige, kutan-subkutane Infiltrate, in der Umgebung können weitere Knoten und Plaques entstehen. Eine Ausbreitung auf die Lymphknoten und Organe ist extrem selten [7]. Vorwiegend tritt die Erkrankung um die 6. Lebensdekade auf (median 59 Jahre).

Das PCFCL ist das häufigste B-Zell-Lymphom der Haut mit häufiger Lokalisation an Kopf, Hals, Nacken und oberem Stammbereich. Eine Ausbreitung auf weitere Organe ist äußerst selten.
Histologisch finden sich unterhalb einer Grenzzone, d. h. von der unauffälligen Epidermis durch eine schmale Zone weitgehend regelhafter Dermis abgesetzt, knotige bis diffuse Infiltrate, die häufig bis in das subkutane Fettgewebe hineinreichen. Die Tumorzellen zeigen eine Differenzierung von Keimzentrumszellen im Sinne von Zentrozyten und Zentroblasten ([Abb. 9]). Das Wachstumsmuster ist variabel und u. a. abhängig von der Tumorlokalisation und dem Alter der einzelnen Läsion. Im Kopfhautbereich sowie in frühen Läsionen findet sich häufig ein sog. follikuläres Wachstumsmuster mit Ausbildung von neoplastischen Follikeln. Weiter fortgeschrittene Läsionen weisen dagegen häufiger ein diffuses Wachstumsmuster auf. Insbesondere Fälle mit diffusem Muster und Prädominanz von großen Zentro-/Immunoblasten gilt es differenzialdiagnostisch vom PCBLT abzugrenzen. Gelegentlich können aber auch beide Muster, also ein follikuläres und diffuses Wachstumsmuster, nebeneinander beobachtet werden. Initiale Läsionen können einen beträchtlichen Anteil reaktiver T-Zellen aufweisen, die im Zuge des Tumorwachstums mit zunehmender Anzahl und Größe neoplastischer B-Zellen kontinuierlich an Zahl abnehmen. Eine prominente Mitreaktion des Stromas ist typisch. Charakteristisch für neoplastische Follikel sind eine reduzierte bis fehlende Mantelzone, ein monomorphes Zellbild mit Aufhebung von hellen und dunklen Zonen sowie eine im Vergleich zu reaktiven Follikeln deutlich reduzierte Mitoserate (< 50 %). Immunhistologisch zeigen sich die neoplastischen Zellen positiv für CD20, CD79a, Bcl-6 sowie negativ für CD5, CD43, Mum-1 und in den meisten Fällen auch für Bcl-2 ([Abb. 10]). CD10 wird in der Regel nur in Fällen mit follikulärem Wachstumsmuster exprimiert, wohingegen Fälle mit diffusem Wachstumsmuster meist negativ sind. Bcl-2- und Mum-1-Negativität ist hilfreich für die Abgrenzung des diffusen PCFCL vom diffusen großzelligen PCBLT, das eine deutlich schlechtere Prognose aufweist. Auch eine Abgrenzung vom sekundär kutanen Keimzentrumslymphom ist möglich, da diese in der Regel positiv für Bcl-2 und CD10 sind [8] [9] [10] [11]. Eine klonale Umlagerung des Immunglobulin-Schwerketten-Gens lässt sich mit Hilfe der PCR in der Mehrzahl der Fälle nachweisen.


Prognose: Trotz der häufig auftretenden kutanen Rezidive, die in bis zu 50 % auftreten können, haben die Patienten meist eine exzellente Prognose (5-JÜR 95 %), da eine systemische Manifestation selten vorkommt [12]. Falls Läsionen an den Unterschenkeln beschrieben werden, verschlechtern sich die Prognose und die 5-JÜR auf 41 % [5].
#
Primär kutanes diffuses großzelliges B-Zell-Lymphom des Unterschenkels, „leg type“
Das PCBLT kommt vor allem bei älteren Frauen vor (median 65 Jahre) und tritt meist, aber nicht ausschließlich, am Bein auf. Diese Erkrankung präsentiert sich in der Regel mit schmerzlosen, rasch wachsenden rot-lividen, derben, disseminierten Knoten, die zu Ulzerationen neigen ([Abb. 11]). Die inguinalen Lymphknoten sind häufig vergrößert. Im Verlauf werden tiefe Gewebsschichten erreicht und ein extrakutaner Befall nachgewiesen.

Histologisch findet sich unterhalb einer meist unauffälligen Epidermis ein dichtes, diffuses, monomorphes Infiltrat, das häufig die gesamte Dermis einnimmt, Adnexstrukturen zerstört und bis in das subkutane Fettgewebe hineinreicht. Das Infiltrat besteht überwiegend aus mittelgroßen bis großen Zentroblasten und Immunoblasten. Atypische Mitosefiguren sind häufig ([Abb. 12] und [Abb. 13]). Im Gegensatz zum PCFCL fehlen Zentrozyten. Eine Mitreaktion des Stromas sowie reaktive T-Zellen sind ebenfalls im Vergleich zum PCFCL deutlich weniger vorhanden. Immunhistologisch zeigen sich die neoplastischen Zellen positiv für CD20, CD79a und Bcl-6 sowie negativ für CD5 und CD10. Bcl-2 und Mum-1 sind beim PCBLT vom diffusen Typ deutlich exprimiert, welches sich hierdurch immunhistologisch vom PCFCL unterscheidet ([Abb. 14]) [1]. Eine klonale Umlagerung des Immunglobulin-Schwerketten-Gens lässt sich mit Hilfe der PCR in der Mehrzahl der Fälle nachweisen. Die bei dem primär nodalen diffus großzelligen B-Zell-Lymphom beschriebene Translokation t(14;18) ist in PCBLT nicht vorhanden.



Prognose: Dieses häufig bei älteren Frauen auftretende Lymphom ist mit einer deutlich reduzierten 5-JÜR assoziiert (50 %). Im Vergleich zu den indolenten PCBCL-Varianten kommt es häufiger zur systemischen Ausbreitung. Es zeigte sich hierbei, dass Patienten mit multiplen Hautläsionen eine deutlich schlechtere Prognose als solche mit einer solitären Läsion bei Diagnosestellung aufweisen [5].
#
Therapie der primär kutanen B-Zell-Lymphome
[Tab. 2] zeigt einen Überblick der aktuellen Therapieempfehlungen bei PCBCL. Trotz der guten Prognose kann es sowohl bei den PCFCL als auch PCMZL nach erfolgreich behandelter Erkrankung zu einem Rezidiv von Lymphomknoten an der Haut kommen. Als First-Line-Therapie kommt eine chirurgische Entfernung oder eine lokale Strahlentherapie infrage (Röntgenweichstrahltherapie 6 – 10 × 2 Gy; 30 – 50 kV, 2 ×/Woche, schnelle Elektronen 40 Gy) [14]. Da ein Zusammenhang von Borrelia burgdorferi-Infektionen mit PCMZL beschrieben wurde, sollte bei Nachweis eines IgG- oder IgM-Titers als Therapie der 1. Wahl eine Antibiotikatherapie, z. B. mit Doxycyclin oder Ceftriaxon erfolgen [15]. Beim Vorliegen mehrerer Herde ist die Injektion mit dem Kortikoid Triamcinolon in die Lymphomknoten möglich. Alternativ sind Interferon-alpha oder Rituximab, ein gegen das B-Zell-Antigen CD20 gerichteter humanisierter Antikörper, zur Behandlung geeignet [16]. Die beiden letztgenannten können entweder direkt in betroffene Hautstellen [17] oder als subkutane Injektion (IFNa) bzw. intravenöse Infusion (Rituximab) gegeben werden [18]. Bei Nachweis von extrakutanem Befall ist primär eine intravenöse Gabe von Rituximab ggf. in Kombination mit einer Chemotherapie angezeigt.
Aufgrund des ähnlich aggressiven Verlaufs sollten PCBLT analog zu den systemischen diffus großzelligen B-Zell-Lymphomen behandelt werden.
Zur Behandlung von PCBLT kommen in erster Linie die intravenöse Gabe von Rituximab in Kombination mit einer Polychemotherapie aus Cyclophosphamid, Hydroxydaunorubicin (Doxorubicin), Oncovin (Vincristin) und Prednisolon (R-CHOP) in Betracht [19]. R-CHOP zeigte sich hierbei gegenüber dem alleinigen Einsatz von CHOP bezüglich des rezidivfreien Intervalls und der Gesamtüberlebensrate signifikant überlegen [20].
Häufig ist allerdings eine Therapie mit R-CHOP limitiert, insbesondere aufgrund der hohen Nebenwirkungsrate des Doxorubicins. Unter einer Therapie mit Doxorubicin kann es zu einer kumulativ dosis-abhängigen dilatativen Kardiomyopathie [21] kommen. Weiterhin sollten bei einer Therapie mit R-CHOP häufig bestehende Komorbiditäten der älteren Patienten berücksichtigt und gegebenenfalls die Dosis angepasst werden. Falls eine Therapie mit R-CHOP kontraindiziert ist, kann wahlweise eine Second-Line-Therapie mit liposomalen Doxorubicin, welches mit oder ohne Polyethylenglykol beschichtet ist, in einer Dosierung von 20 mg/m2 Körperoberfläche eingesetzt werden [22] [23] [24]. Aufgrund der liposomalen Carrier ist eine höhere Dosierungsmöglichkeit bei gleichzeitig verringerter Nebenwirkungsrate möglich. Auch das Pyrimidinanologon Gemcitabin kann in der Second-Line-Therapie mit Dosierungen bis 1200 mg/m2 in Betracht gezogen werden. Alternativ kann bei umschriebenem Befund und Gründen, die gegen eine innerliche Therapie sprechen, eine Strahlentherapie durchgeführt werden [25].
Zur Behandlung der PCBCL sind rekombinantes Interferon-α-2a, Steroid sowie Rituximab als intraläsonales Therapeutika zugelassen. Polychemotherapien mit CHOP können in Kombination mit Rituximab als R-CHOP-Therapie eingesetzt werden. Als Second-Line-Therapie kommen Monochemotherapeutika zum Einsatz.
#
Ausblick
Neben den oben genannten Standardtherapien zur Therapie der PCBCL befinden sich zahlreiche neue Therapien in der Entwicklung und in Zulassungsstudien. Hier ist u. a. der mit Yttrium-90 markierte, monoklonale CD20-IgG1-Antikörper [90Y] Ibritumomab-Tiuxetan zu nennen [26]. Maza et al. sahen eine 100 %-Ansprechrate bei PCBCL und eine mittlere Remissionsdauer von 12 Monaten. Aufgrund auftretender Nebenwirkungen insbesondere einer Myelosuppression ist diese Therapie nur beim PCBLT zu empfehlen, falls es bei den o. g. Standardtherapien nicht zu einem adäquaten Therapieansprechen kommt. Aber auch die neuen CD20-Antikörper wie z. B. Ofatumumab zeigen eine deutlich höhere Bindungsaffinität, was sich klinisch bei systemischen B-Zell-Lymphomen in einem deutlich höheren Ansprechen zeigt und somit auch für die aggressiven kutanen B-Zell-Lymphome von höchstem Interesse sein könnte [27].
-
PCBCL stellen eine heterogene Gruppe von Entitäten dar, die sich hinsichtlich der Klinik, der Histologie, des Immunphänotyps und des Genotyps unterscheiden.
-
Die PCBCL-Therapie beruht auf einer exakten Einordnung der klinischen Entitäten anhand der aktuellen WHO-Klassifikation.
-
Die Prognose der indolenten PCBCL ist, bis auf wenige Ausnahmen, exzellent.
-
Kurative Therapieansätze sind beim PCBLT zur Zeit nicht vorhanden. Daher zielt die Therapie auf die Verbesserung der Symptomatik und Lebensqualität. Hierzu stehen sowohl intraläsionale, strahlentherapeutische als auch systemische Therapien zur Verfügung.
-
Als intraläsionale Therapeutika sind zur Behandlung der PCBCL rekombinantes Interferon-α-2a, Steroid sowie Rituximab zugelassen. Polychemotherapien mit CHOP können in Kombination mit Rituximab als R-CHOP-Therapie eingesetzt werden.
-
Monochemotherapeutika, wie z. B. Gemcitabin und das liposomal verkapselte Doxorubicin, kommen als Second-Line-Therapie zum Einsatz.
#
#
Interessenkonflikt
Die Autoren geben an, dass kein Interessenkonflikt besteht.
-
Literatur
- 1 Willemze R, Jaffe ES, Burg G et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105: 145-152
- 2 Swerdllow SH, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2008
- 3 Bailey EM, Ferry JA, Harris NL et al. Marginal zone lymphoma (low-grade B-cell lymphoma of mucosa associated lymphoid tissue type) of skin and subcutaneous tissue. Am J Surg Pathol 1996; 20: 1011-1023
- 4 Sandberg Y, Heule F, Lam K et al. Molecular immunoglobulin/T-cell receptor clonality analysis in cutaneous lymphoproliferations. Experience with the BIOMED-2 standardized polymerase chain reaction protocol. Haematologica 2003; 88: 659-670
- 5 Senff NJ, Noordijk EM, Kim YH et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 2008; 112: 1600-1609
- 6 Ziemer M, Bauer HI, Fluhr JW et al. Primary cutaneous follicle center lymphoma-crosti lymphoma: what can we learn?. Am J Clin Dermatol 2008; 9: 133-136
- 7 Garcia CF, Weiss LM, Warnke RA et al. Cutaneous follicular lymphoma. Am J Surg Pathol 1986; 10: 454-463
- 8 Cerroni L, Volkenandt M, Rieger E et al. Bcl-2 protein expression and correlation with the interchromosomal 14;18 translocation in cutaneous lymphomas and pseudolymphomas. J Invest Dermatol 1994; 102: 231-235
- 9 Child FJ, Russell-Jones R, Woolford AJ et al. Absence of the t(14;18) chromosomal translocation in primary cutaneous B-cell lymphoma. Br J Dermatol 2001; 144: 735-744
- 10 Schreuder MI, Hoefnagel JJ, Jansen PM et al. FISH analysis of MALT lymphoma-specific translocations and aneuploidy in primary cutaneous marginal zone lymphoma. J Pathol 2005; 205: 302-310
- 11 Streubel B, Lamprecht A, Dierlamm J et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 2003; 101: 2335-2339
- 12 Pimpinelli N, Santucci M, Bosi A et al. Primary cutaneous follicular centre-cell lymphoma – a lymphoproliferative disease with favourable prognosis. Clin Exp Dermatol 1989; 14: 12-19
- 13 Kodama K, Massone C, Chott A et al. Primary cutaneous large B-cell lymphomas: clinicopathologic features, classification, and prognostic factors in a large series of patients. Blood 2005; 106: 2491-2497
- 14 Stadler R, Assaf C, Klemke CD et al. Brief S2k guidelines Cutaneous lymphomas. J Dtsch Dermatol Ges 2013; 11: 19-28
- 15 Cerroni L, Zochling N, Putz B et al. Infection by Borrelia burgdorferi and cutaneous B-cell lymphomas. J Cutan Pathol 1997; 24: 457-461
- 16 Gellrich S, Muche JM, Wilks A et al. Systemic eight cycle anti-CD20 monoclonal antibody (rituximab) therapy in primary cutaneous B-cell lymphomas-an applicational observation. Br J Dermatol 2005; 153: 167-173
- 17 Heinzerling L, Dummer R, Kempf W et al. Intralesional therapy with anti-CD20 monoclonal antibody Rituximab in primary B-cell lymphoma. Arch Dermatol 2000; 136: 374-378
- 18 Heinzerling L, Urbanek M, Funk JO et al. Reduction of tumor burden and stabilisation of disease by systemic therapy with Anti-CD20 antibody (Rituximab) in patients with primary cutaneous B-cell lymphoma. Cancer 2000; 89: 1835-1844
- 19 Coiffier B, Lepage E, Brière J et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large B-cell lymphoma. N Engl J Med 2002; 346: 235-242
- 20 Habermann TM, Weller EA, Morrison VA et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 2006; 24: 3121-3127
- 21 Chatterjee K, Zhang J, Honbo N et al. Doxorubicin cardiomyopathy. Cardiology 2010; 115: 155-162
- 22 Fabbri A, Cencini E, Alterini R et al. Rituximab plus liposomal pegylated doxorubicin in the treatment of primary cutaneous B‐cell lymphomas. European journal of haematology 2014; 93: 129-136
- 23 Pulini S, Rupoli S, Goteri G et al. Efficacy and safety of pegylated liposomal doxorubicin in primary cutaneous B‐cell lymphomas and comparison with the commonly used therapies. European journal of haematology 2009; 82: 184-193
- 24 Assaf C, Becker JC, Beyer M et al. Treatment of advanced cutaneous T-cell lymphomas with non-pegylated liposomal doxorubicin consensus of the lymphoma group of the Working Group Dermatologic Oncology. J Dtsch Dermatol Ges 2013; 11: 338-347
- 25 Senff NJ et al. Results of radiotherapy in 153 primary cutaneous B-Cell lymphomas classified according to the WHO-EORTC classification. Arch Dermatol 2007; 143: 1520-1526
- 26 Maza S, Gellrich S, Assaf C et al. Yttrium-90 ibritumomab tiuxetan radioimmunotherapy in primary cutaneous B-cell lymphomas: first results of a prospective, monocentre study. Leuk Lymphoma 2008; 49: 1702-1709
- 27 Jabbour E, O’Brien S, Ravandi F et al. Monoclonal antibodies in acute lymphoblastic leukemia. Blood 2015; 125: 4010-4016
Korrespondenzadresse
-
Literatur
- 1 Willemze R, Jaffe ES, Burg G et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105: 145-152
- 2 Swerdllow SH, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2008
- 3 Bailey EM, Ferry JA, Harris NL et al. Marginal zone lymphoma (low-grade B-cell lymphoma of mucosa associated lymphoid tissue type) of skin and subcutaneous tissue. Am J Surg Pathol 1996; 20: 1011-1023
- 4 Sandberg Y, Heule F, Lam K et al. Molecular immunoglobulin/T-cell receptor clonality analysis in cutaneous lymphoproliferations. Experience with the BIOMED-2 standardized polymerase chain reaction protocol. Haematologica 2003; 88: 659-670
- 5 Senff NJ, Noordijk EM, Kim YH et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 2008; 112: 1600-1609
- 6 Ziemer M, Bauer HI, Fluhr JW et al. Primary cutaneous follicle center lymphoma-crosti lymphoma: what can we learn?. Am J Clin Dermatol 2008; 9: 133-136
- 7 Garcia CF, Weiss LM, Warnke RA et al. Cutaneous follicular lymphoma. Am J Surg Pathol 1986; 10: 454-463
- 8 Cerroni L, Volkenandt M, Rieger E et al. Bcl-2 protein expression and correlation with the interchromosomal 14;18 translocation in cutaneous lymphomas and pseudolymphomas. J Invest Dermatol 1994; 102: 231-235
- 9 Child FJ, Russell-Jones R, Woolford AJ et al. Absence of the t(14;18) chromosomal translocation in primary cutaneous B-cell lymphoma. Br J Dermatol 2001; 144: 735-744
- 10 Schreuder MI, Hoefnagel JJ, Jansen PM et al. FISH analysis of MALT lymphoma-specific translocations and aneuploidy in primary cutaneous marginal zone lymphoma. J Pathol 2005; 205: 302-310
- 11 Streubel B, Lamprecht A, Dierlamm J et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 2003; 101: 2335-2339
- 12 Pimpinelli N, Santucci M, Bosi A et al. Primary cutaneous follicular centre-cell lymphoma – a lymphoproliferative disease with favourable prognosis. Clin Exp Dermatol 1989; 14: 12-19
- 13 Kodama K, Massone C, Chott A et al. Primary cutaneous large B-cell lymphomas: clinicopathologic features, classification, and prognostic factors in a large series of patients. Blood 2005; 106: 2491-2497
- 14 Stadler R, Assaf C, Klemke CD et al. Brief S2k guidelines Cutaneous lymphomas. J Dtsch Dermatol Ges 2013; 11: 19-28
- 15 Cerroni L, Zochling N, Putz B et al. Infection by Borrelia burgdorferi and cutaneous B-cell lymphomas. J Cutan Pathol 1997; 24: 457-461
- 16 Gellrich S, Muche JM, Wilks A et al. Systemic eight cycle anti-CD20 monoclonal antibody (rituximab) therapy in primary cutaneous B-cell lymphomas-an applicational observation. Br J Dermatol 2005; 153: 167-173
- 17 Heinzerling L, Dummer R, Kempf W et al. Intralesional therapy with anti-CD20 monoclonal antibody Rituximab in primary B-cell lymphoma. Arch Dermatol 2000; 136: 374-378
- 18 Heinzerling L, Urbanek M, Funk JO et al. Reduction of tumor burden and stabilisation of disease by systemic therapy with Anti-CD20 antibody (Rituximab) in patients with primary cutaneous B-cell lymphoma. Cancer 2000; 89: 1835-1844
- 19 Coiffier B, Lepage E, Brière J et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large B-cell lymphoma. N Engl J Med 2002; 346: 235-242
- 20 Habermann TM, Weller EA, Morrison VA et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol 2006; 24: 3121-3127
- 21 Chatterjee K, Zhang J, Honbo N et al. Doxorubicin cardiomyopathy. Cardiology 2010; 115: 155-162
- 22 Fabbri A, Cencini E, Alterini R et al. Rituximab plus liposomal pegylated doxorubicin in the treatment of primary cutaneous B‐cell lymphomas. European journal of haematology 2014; 93: 129-136
- 23 Pulini S, Rupoli S, Goteri G et al. Efficacy and safety of pegylated liposomal doxorubicin in primary cutaneous B‐cell lymphomas and comparison with the commonly used therapies. European journal of haematology 2009; 82: 184-193
- 24 Assaf C, Becker JC, Beyer M et al. Treatment of advanced cutaneous T-cell lymphomas with non-pegylated liposomal doxorubicin consensus of the lymphoma group of the Working Group Dermatologic Oncology. J Dtsch Dermatol Ges 2013; 11: 338-347
- 25 Senff NJ et al. Results of radiotherapy in 153 primary cutaneous B-Cell lymphomas classified according to the WHO-EORTC classification. Arch Dermatol 2007; 143: 1520-1526
- 26 Maza S, Gellrich S, Assaf C et al. Yttrium-90 ibritumomab tiuxetan radioimmunotherapy in primary cutaneous B-cell lymphomas: first results of a prospective, monocentre study. Leuk Lymphoma 2008; 49: 1702-1709
- 27 Jabbour E, O’Brien S, Ravandi F et al. Monoclonal antibodies in acute lymphoblastic leukemia. Blood 2015; 125: 4010-4016