

Subscribe to RSS
DOI: 10.1055/s-0033-1340073
Strategies for the Synthesis of Cyclic Ethers of Marine Natural Products
Authors
Publication History
Received: 31 July 2013
Accepted after revision: 24 September 2013
Publication Date:
26 November 2013 (online)

Abstract
This account describes our studies on the synthesis of natural products that contain cyclic ethers in their structures. An overview of the main methodologies is presented and several total syntheses developed by the group are described. We also discuss new applications based on the use of Prins and Nicolas reactions as key steps in the preparation of oxygenated heterocyclic compounds.
1 Introduction
1.1 Natural Sources and Biogenesis
2 Carbon–Oxygen Bond-Formation Approach
2.1 Intramolecular Opening of Epoxy Alcohols and Related Bromo Cyclizations
2.2 Intramolecular Hetero-Michael Reaction of Alkoxy γ-Benzoyloxy α,β-Unsaturated Esters
2.3 Intramolecular Nicholas Reaction
3 Ring-Closing Metathesis of Unsaturated Linear Ethers
4 Prins Cyclization Promoted by Iron(III) salts
4.1 General Strategy
4.2 Cyclization Using Alkynols and the Formation of Vinyl Halides
4.3 The Use of Trimethylsilyl and Electron-Withdrawing Substituents in the Synthesis of 2,6-Disubstituted Tetrahydropyrans
4.4 Catalytic Prins Cyclization
4.5 Synthesis of Oxepanes
5 Summary
Biographical Sketches

Tomás Martín received his B.Sc. in chemistry from the University of La Laguna, Tenerife, Spain in 1991, and obtained his Ph.D. in 1996 from the same university, working on the enantioselective synthesis of γ-lactones under the guidance of Dr. C. M. Rodríguez and Professor Víctor S. Martín. He was a postdoctoral fellow of the Spanish Ministry of Education and Science (MEC) at the Scripps Research Institute from January 1997 to December 1998, working on molecular recognition under the supervision of Professor Julius Rebek. In 1999, he returned to the Canary Islands, with a reincorporation contract of the Spanish MEC, and joined Victor S. Martín´s group. In 2002, he won a contract on the Ramón y Cajal program, created to boost Spanish science. In 2005, he became a tenured scientist at the Instituto de Productos Naturales y Agrobiología-CSIC. His research interest includes supramolecular chemistry, methodology, and total synthesis of bioactive natural products.

Juan I. Padrón received his B.Sc. in chemistry from the University of La Laguna, Tenerife, Spain in 1990, and obtained his Ph.D. in organic chemistry in 1995 from the same university, working on the synthesis and stereochemical studies of saccharides under the guidance of Professor Jesús Trujillo Vázquez. He then spent two years (1996–1997) at the University of Columbia as a postdoctoral fellow with Professor Samuel J. Danishefsky, working on natural product synthesis. In 1998, he returned to the Canary Islands, and joined the laboratory of Professor Jaime Bermejo (CSIC). In 2004, he won a contract under the Ramón y Cajal program, created to boost Spanish science, working with Víctor S. Martín at the University of La Laguna. In 2008, he became a tenured scientist at the Instituto de Productos Naturales y Agrobiología-CSIC, Tenerife, Spain. His research interests include sustainable metal catalysis and new reaction methodologies and their applications to natural product synthesis.

Víctor S. Martín was born in La Palma (Canary Islands), Spain, in 1953 and gained his Ph.D. degree at the University of La Laguna, Tenerife, in 1978 under the guidance of Professors Antonio González, Julio D. Martín, and Manuel Norte. From 1980 to 1982, he was a postdoctoral fellow with Professor K. Barry Sharpless at Stanford University and at the Massachusetts Institute of Technology. He moved to the University of La Laguna as an associate professor in 1984. In 1988, was promoted to scientific researcher of the CSIC of Spain. Since 1992, he has held a full professorial position at the University of La Laguna. His research interests include asymmetric synthesis, methodology, and total synthesis of bioactive natural products.
Introduction
Polyfunctionalized cyclic ethers are the structural keystones of a wide range of biologically active natural products, including polyether antibiotics,[1] ladder marine toxins,[2] annonaceous acetogenins,[3] and lauroxanes[4] (Figure [1]). Many synthetic chemists have made extensive studies on the development of new strategies and efficient methodologies for the construction of ether ring systems and their applications in total syntheses of natural polycyclic ethers. A number of excellent reviews on synthetic aspects of naturally occurring cyclic ethers have been published.[5]
In the development of methodologies for constructing cyclic ethers, three major approaches have been adopted; these involve (a) final C–O bond formation using a nucleophilic oxygen, (b) C–C bond formation in a preconstructed linear ether, or (c) simultaneous C–O and C–C bond formation in a single step (Scheme [1]).


Approach (a) is usually based on attack by a nucleophilic oxygen (generally a hydroxy or epoxy group) on an activated functional group (an epoxide, a leaving group on a saturated carbon, or an electrophilically activated alkene).[6] The synthesis of cyclic ethers from lactones can be considered to fall within this category.[7] Methods based on C–C bond formation [approach (b)] involve the formation of an acyclic ether that undergoes subsequent cyclization. Reactions such as ring-closing metathesis (RCM)[8] or samarium-based coupling of aldehydes with suitable electrophilic alkenes have been applied to this end.[5a] Finally, the possibility of creating the oxygenated ring system by a process involving the formation of both a C–O bond and a C–C bond [Approach (c)] is highly desirable. Among processes of this type, those based on the Prins cyclization have attracted a great deal of attention.[9]
With the aim of develop methodologies for the synthesis of bioactive natural products, especially those of marine origin, we have explored all three approaches outlined above. An overview of our researches in the synthesis of cyclic ethers is presented in this account.
1.1Natural Sources and Biogenesis
Because of our interest in the natural products described in this account, we will summarize some of the most representative examples of biogenesis. As we will see, these biogenetic ideas have often been a source of inspiration for the development of methodologies used in syntheses of various natural products.
Red and brown algae are very important sources of marine natural products that exhibit a wide range of biological properties, including antitumor, antimicrobial, immunosuppressant, antifeedant, and pesticidal activities.[10] Red algae of the genus Laurencia produce many unique compounds, including a series of nonterpenoidal C15-metabolites, generally named lauroxanes, derived from metabolism of C16 fatty acids (acetogenins).[10] For example, linear compounds, such the enantiomers (3E,9Z,12E)-pentadeca-3,9,12-trien-1-yne-6,7-diol [trans-laurediol, (E)-6] and (3Z,9Z,12E)-pentadeca-3,9,12-trien-1-yne-6,7-diol [cis-laurediol, (Z)-6],[11] and cyclic products such as trans-(–)-kumausyne (4) and trans-(+)-deacetylkumausyne (5) have been isolated from Laurencia nipponica.[12] Lauroxanes all contain polysubstituted cyclic ether moieties with a defined substituent stereochemistry and a ring size ranging from five to nine atoms. These cyclic ethers are considered to originate biogenetically from laurediols (6) through intramolecular electrophilic cyclizations induced by the bromonium ion (Scheme [2]). Biosynthesis studies performed by Murai and co-workers[13] demonstrated that lactoperoxidase and bromoperoxidase (BPO) catalyze this bromocyclization.

On the other hand, (2S,3S,5R)-5-[(1R)-1-hydroxydec-9-en-1-yl]-2-pentyltetrahydrofuran-3-ol (7)[14] and (2S,3S,5S)-5-[(1S)-1-hydroxydec-9-en-1-yl]-2-pentyltetrahydrofuran-3-ol (8)[15] are C19-diols isolated from Notheia anomala, a member of the Notheiaceae family, a taxonomically unique brown alga found along the coasts of southern Australia. These tetrahydrofurans are believed to be biosynthesized through cyclization of the natural methylene-interrupted bisepoxides 9 and 10, respectively (Scheme [3]).[16] They are potent and selective inhibitors of the larval development of parasitic nematodes.[14]

A group of halogenated triterpenoids with several levels of oxidation and various cyclic systems form an important class of natural products isolated from Laurencia. All known metabolites are squalene-derived noncarbocyclic polyethers formed from the corresponding squalene-derived polyepoxides (Scheme [4]).[17] We became particularly interested in the synthesis of teurilene (11) and thyrsiferol (12) and its congeners. Teurilene (11) was isolated, along with thyrsiferol (12) and its acetyl derivative thyrsiferyl 23-acetate (13), from the red alga L. obtusa.[18] Despite of containing eight stereogenic centers and three tetrahydrofuran rings in its structure, teurilene has a meso symmetry (Cs ) making it an achiral compound. Thyrsiferol (12) was the first example of a marine squalene-derived polyether triterpenoid containing a dioxabicyclo[4.4.0]decane moiety (the B–C ring). This compound was isolated by Blunt et al. from L. thyrsifera collected off New Zealand.[19]

Polyether ladder toxins produced by dinoflagellates form one of the most spectacular groups of marine polyethers in terms of both their structures and their biological activities. This group includes compounds such as ciguatoxin,[20] brevetoxin A (3),[21] brevetoxin B (14),[22] maitotoxin,[23] and yessotoxin.[24] Structurally, these highly toxic polycyclic toxins are complex molecules consisting of fused cyclic ether units, usually with a trans relationship between the two substituents (H or CH3) at the point of ring fusion, and a syn stereochemistry of substituents (H or CH3) close to the oxygen atom of the ring. These compounds are extremely toxic and are responsible for neurotoxic shellfish poisoning, diarrheic shellfish poisoning, ciguatera, and other illnesses. It is accepted that these ladder toxins might be biosynthesized through a polyepoxide cascade cyclization (Scheme [5]).[25]

Natural products that contain cyclic ethers are not exclusive to the marine environment. For instance, polyether ionophores are a large group of natural biologically active substances produced by Streptomyces species. They are lipid-soluble and can transport metal cations across cell membranes.[1] Another very interesting group of cyclic ethers from terrestrial environments are the polyether annonaceous acetogenins.[3] [26] This group of substances are natural products isolated exclusively from Annonaceae species, which are widely distributed in tropical and subtropical regions. The typical skeleton of these substances is generally characterized by an unbranched C32 or C34 fatty acid ending in a butenolide moiety. Structurally, the compounds can be monotetrahydrofurans, adjacent bistetrahydrofurans, nonadjacent bistetrahydrofurans, tertetrahydrofurans, adjacent tetrahydrofuran–tetrahydropyrans, nonadjacent tetrahydrofuran–tetrahydropyrans, monotetrahydropyrans, or exclusively γ-lactones. The compounds have attracted the attention of many research groups because of their interesting structural complexity and their important biological activities (anticancer, antiprotozoal, pesticide, immunosuppressive, and antifeedant properties).[26] Biogenetically, the annonaceous acetogenins appear to be formed through the polyketide pathway. The tetrahydrofuran, tetrahydropyran, and epoxide rings might arise from isolated double bonds that undergo epoxidation and cyclization (Scheme [6]).[27]

2
Carbon–Oxygen Bond-Formation Approach
This approach represents the tactic that most closely resembles the way that nature synthesizes cyclic ethers. We will start by examining those methods and their applications in the synthesis of marine natural products that contain tetrahydrofuran moieties as their main structural features. The difficulties in extending such methods to the synthesis of tetrahydropyrans prompted us to develop alternative approaches for synthesizing larger cyclic ethers. The intramolecular O-hetero Michael addition of a hydroxy group to a γ-substituted α,β-unsaturated ester and a Nicholas reaction using a hydroxy or epoxide group have emerged as robust methodologies in this respect.
2.1Intramolecular Opening of Epoxy Alcohols and Related Bromo Cyclizations
Within our program directed toward the stereoselective total synthesis of bioactive substances of marine origin,[28] we focused our attention on metabolites 4, 5, 7, and 8, which contain highly functionalized tetrahydrofuran rings (Scheme [7]). Essentially, our idea was to synthesize the tetrahydrofuran ring by an intramolecular opening of an epoxide or bromonium derivative available from a suitable alkene. We surmised that the necessary linear unsaturated precursors might be stereoselectively synthesized by Wittig-type reactions of the lactol 16; this, in turn, would be accessible from the key γ-lactone 17. Lactone 17 should, in turn, be obtainable in both enantiomeric forms by asymmetric dihydroxylation of the β,γ-unsaturated ester 18.[29]

The necessary β,γ-unsaturated esters 18 were synthesized from the corresponding aldehydes by a modified Knoevenagel condensation with malonic acid in the presence of piperidine as catalyst under nonpolar conditions, as exemplified in Scheme [8].[30] Sharpless asymmetric dihydroxylation with the appropriate AD-mix gave the chiral β-hydroxy γ-lactone 17a. In this step, we simultaneously achieved two important goals: first, we introduced the correct stereochemistry at two stereocenters for the final molecule, and secondly, by the lactonization, we differentiated chemically between the two hydroxy groups. Protection of the free secondary alcohol gave 17b, which underwent selective reduction with diisobutylaluminum hydride to give the corresponding lactols, primed for subsequent Wittig homologation. For the stereoselective synthesis of the C19-dihydroxy tetrahydrofurans 7 and 8, we considered a biomimetic approach based on the cyclization of the enantiomerically enriched bisepoxides 9 and 10.[31] However, this approach would involve stereoselective synthesis of epoxides derived from Z-olefins which, in general terms, is difficult to achieve. [31] [32] To overcome this difficulty, we focused our attention on the terminal 2,3-epoxy alcohol 20, which has the correct stereochemistry and is easily available by a Katsuki–Sharpless asymmetric epoxidation of the corresponding E-allylic alcohol 19, obtained by homologation of the protected β-hydroxy γ-lactone 17b. This strategy has three major advantages. First, the synthesis of the diastereoisomer 22 can be achieved simply by changing the chiral auxiliary at the asymmetric epoxidation step. Secondly, a concomitant cyclization to the tetrahydrofuran ring occurs during the epoxidation step. Thirdly, the application of two enantioselective reactions ensures that the final products have high enantiomeric purities.[33]

The existence of precedents relative to the total synthesis of trans-kumausyne from dihydroxy tetrahydrofurans similar to 23 prompted us to perform a short formal synthesis of a lauroxane (Scheme [9]).[34] This started with butane-1,4-diol. The application of a series of steps similar to those used in the syntheses of 21 and 23 gave tetrahydrofuran 24 as a pure stereoisomer. Protection of the 1,2-diol functionality as a dimethylacetal followed by cleavage of the benzyl ether and subsequent oxidation gave the corresponding aldehyde, which, without purification, was treated with the ylide derived from commercially available triphenyl[3-(trimethylsilyl)prop-2-yn-1-yl]phosphonium bromide to give the trans-enyne 25 as the sole geometrical isomer. Removal of the silyl groups, acetylation of the secondary alcohol, hydrolysis of the dimethyl acetal, and oxidative cleavage of the diol gave the aldehyde 26. Because this aldehyde had previously been transformed into trans-(–)-kumausyne (4),[34] our approach constituted a new formal synthesis of this natural product.[35]

In view of the simplicity with which it is possible to obtain vicinal diols stereoselectively, we considered that the methodology described above might also be used for the synthesis of laurediols 6 (Scheme [10]). We started with the protected lactone 17d, which we subjected to carbon homologation in both directions to give the desired framework. Removal of the benzyl protecting group, oxidation of the primary alcohol, and stereoselective Wittig reaction provided the (Z)-olefin 27. To complete the synthesis, we reduced the γ-lactone with one equivalent of diisobutylaluminum hydride, and submitted the resulting lactol to Wittig olefination with the ylide derived from commercially available triphenyl[3-(trimethylsilyl)prop-2-yn-1-yl]phosphonium bromide to give the trans-enyne. Fluoride-induced cleavage of the silyl-protecting groups then gave trans-(+)-laurediol (trans-6).[36]

For the synthesis of exo-bromotetrahydrofurans, we considered an approach that is formally similar to the biogenetic origin of this type of compound.[28c] Basically, this involved the synthesis of the tetrahydrofuran ring by an electrophilic bromocyclization of a suitable linear precursor. Because laurediols are assumed to be biogenetic precursors of the whole series of compounds (see above), a quite logical extension of our work was the direct synthesis of lauroxanes by this type of reaction (Scheme [11]). Thus, when we submitted trans-(+)-laurediol (trans-6) to a bromocyclization reaction with 2,4,4,6-tetrabromocyclohexa-2,5-dien-1-one, we obtained trans-(+)-deacetylkumausyne (5), albeit with poor stereoselectivity.[37]

The low stereoselectivity achieved in the cyclization step prompted us to study the factors that might influence the stereochemical course of the reaction. Such studies might have eventually led to an alternative approach to the synthesis of the natural compounds. We therefore planned a different approach to (+)-deacetylkumausyne (5) based on the retrosynthetic analysis outlined in Scheme [12]. We attempted to achieve high convergence in the synthesis of the hydroxyalkene precursors, with the aim of scaling up the amount of final product that was produced.

We began our synthesis from the silyl-protected threo-1,2-epoxy-3-alcohol 29, readily available from the known enyne 28 (Scheme [13]).[38] The silyl epoxy ether 29 was used as the central fragment for homologation of the chain in both directions. Thus, the double bond in 29 was cleaved with ozone under reductive conditions and the resulting aldehyde was submitted to the appropriate Wittig reaction to give diene 30 with excellent stereocontrol. The terminal epoxide was opened with the lithium salt of a protected propargylic alcohol to give the linear chain 31, in which all the hydroxy groups were chemically differentiated ready for the cyclization reaction.[39] When we treated a solution of alkyne 31 in dichloromethane with 2,4,4,6-tetrabromocyclohexa-2,5-dien-1-one as a source of bromonium ions, we obtained a 1:1 mixture of the tetrahydrofurans 32 and 33 after cleavage of the tetrahydropyran protecting group.

Although the above reaction showed better stereoselectivity, we considered the possibility of improving this further by manipulating the stereochemistry of the silyl-protected carbinol. We therefore prepared the precursor 35 from the erythro-epoxy ether 34 by a similar sequence of reactions (Scheme [14]). The bromocyclization step gave the desired syn-2,5-disubstituted tetrahydrofuran 36 with an enhanced selectivity of 5:1. The one-carbon homologation to the trans-enyne was performed by Corey’s method.[40] Finally, inversion of the configuration of the secondary carbinol by a consecutive sequence of oxidation and reduction reactions gave trans-(+)-deacetylkumausyne (5).[41]

2.2
Intramolecular Hetero-Michael Reaction of Alkoxy γ-Benzoyloxy α,β-Unsaturated Esters
Intramolecular cleavage of an epoxide by an internal oxygen nucleophile favors the smaller heterocycle as a result of a transition state in which the attacking oxygen and the breaking oxirane C–O bond are collinear (exo-opening).[42] In most approaches for promoting the desired endo-cleavage directing groups must be covalently attached to the epoxides.[43] Water can serve as a promoter of an endo-selective epoxide-opening cascade if a single templating tetrahydropyran moiety is appended to a chain of epoxides.[44]
The intramolecular Michael addition of an oxygen atom to an α,β-unsaturated ester can be considered as an interesting alternative strategy for the synthesis of tetrahydropyran rings. We considered the possibility that the geometry of the double bond might play an important role in the transition state of this hetero-Michael addition. Such a synthetic approach can be considered as being based on the possible biogenetic route to some natural tetrahydropyran systems. In this respect, it had been proposed that the tetrahydropyran ring of the polyketide cladosporin is formed by an intramolecular hetero-Michael addition of a hydroxy group to a suitable α,β-unsaturated carbonyl group.[45]
Our research focused on the study of the effects of the stereochemistry of the chiral centers and the double-bond geometry on the cyclization of substituted chiral 7-hydroxy-4-benzoyloxy-2,3-unsaturated esters (Scheme [15]).[46] We began our studies with the linear models 39 and 40, which have a known absolute configuration of the secondary allylic center. Asymmetric induction was performed by means of the Katsuki–Sharpless asymmetric epoxidation[31] of a suitable allylic alcohol obtained from butane-1,4-diol. The enantiomerically enriched epoxy alcohol 37 was regioselectively opened by using benzoic acid as a nucleophile and titanium tetraisopropoxide as an acidic catalyst to give the benzoyloxy diol with excellent regioselectivity (>100:1).[47] Oxidative cleavage with sodium periodate gave the aldehyde 38, primed for Wittig homologation. Treatment of aldehyde 38 with the sodium salt of trimethyl phosphonoacetate gave the E-unsaturated ester with excellent E/Z selectivity (>20:1). Similarly, when aldehyde 38 was treated with the sodium salt of [2-(diphenoxymethoxy)-2-oxoethyl]phosphonate, we obtained the Z-ester almost exclusively (Z/E >20:1).[48] Cleavage of the tetrahydropyran protecting groups from the E- and Z-esters under standard acidic conditions gave the corresponding free alcohols 39 and 40, respectively. Cyclization of the E-alcohol 39 gave the cis-2,3-disubstituted tetrahydropyran 41, whereas Z-alcohol 40 gave the trans-analogue 42.

A computational study of the above cyclization at the ab initio level showed that coordination between the pre-tetrahydropyranyl oxygen of the carbonyl group of the α,β-unsaturated ester with the cation of the base is necessary to obtain a suitable transition-state model. The experimental results can be rationalized in terms of a model with a pre-chair conformation in which the chiral inductor located in the equatorial position approaches the α,β-unsaturated ester in the axial mode for the E-isomer and in the equatorial mode for the Z-geometry (Scheme [16]).[49]

When the formation of all 2,3,5,6-substituted tetrahydropyrans is considered, the stereochemistry at the C2 position of the ring can be predicted by applying an empirical rule. If the chiral inductor (G) is prevented from adopting an equatorial conformation by geometrical restrictions, the trans-2,3-difunctionalized tetrahydropyran is always obtained. In this case, well-known 1,3-allylic interactions[50] might be the cause of the preferred approach, although a different mechanistic pathway is also possible in such cases.[49]

This method has proved to be excellent for obtaining substituted tetrahydropyrans with high stereochemical purities. Under kinetic control (low temperatures and stoichiometric amounts of a sodium or potassium base), the procedure is particularly efficient for the synthesis of ladder-fused rings (Scheme [18]). Thus, by starting from compounds containing a ring with a well-defined stereochemistry at the carbon on which the nucleophilic hydroxy group was located, six (43–48) of the eight possible diastereoisomers (only one enantiomeric series was considered) were prepared by direct intramolecular hetero-Michael addition reactions. The remaining two stereoisomers (49 and 50) were synthesized by simple chemical transformations of the corresponding diastereoisomers of suitable stereochemistry. It must be emphasized that the isomerization of 46 to 50 was performed under thermodynamic conditions (room temperature). In a similar manner, 44 and 47 were isomerized to 45 and 48, respectively.[51]

2.3
Intramolecular Nicholas Reaction
Although it might be possible to synthesize seven-membered cyclic ethers by an intramolecular O-hetero Michael reaction, the necessity for the precursors to contain certain structural features (such as a Z-double bond) limits the usefulness of this procedure.[52] To overcome this restriction, we developed a general method for preparing medium-sized cyclic ethers (six to nine members) that involves the intramolecular attack by a hydroxy group located on a saturated chain onto a carbocation generated by acid treatment of an exo-(propargyl)hexacarbonyldicobalt complex (the intramolecular Nicholas reaction) (Scheme [19]).[53] [54]

Interestingly, we found that epoxides can also be used as efficient nucleophiles in the intramolecular version of the Nicholas reaction. Thus, intramolecular attack by the epoxide on the exo-cation 52, generated by acid treatment of complex 51, gave the corresponding polysubstituted cyclic ethers (five-, six-, or seven-membered series) in good yields (Scheme [20]). The regioselectivity of the cyclization is affected by a number of factors, including the distance between the epoxide and the propargylic cation, the nature of the protecting group on the primary alcohol, and the reaction conditions.[55]

In a case where competition between the formation of six- or seven-membered rings was possible, a tert-butoxycarbonyl protecting group on the hydroxy group of the terminal 2,3-epoxy alcohol ensured that the disubstituted tetrahydropyran 53 was obtained exclusively (Scheme [21]).

On the basis of this methodology, we performed a retrosynthetic analysis of (+)-muconin (2), in which the tetrahydropyran ring 53 (R = H) would act as a scaffold (Scheme [22]). We first disconnected the saturated chain from the terminal epoxide 54, bearing in mind the possibility of introducing such an alkyl chain by nucleophilic opening of the terminal epoxide ring. The tetrahydrofuran ring might be formed through in situ regio- and stereoselective exo-cyclization of a suitable epoxy alcohol, which, in turn, might be obtained by a Katsuki–Sharpless asymmetric epoxidation of the allylic alcohol 55.[31] This compound could be synthesized by an allyl Grignard regioselective opening of epoxide 56 with further homologation and reduction. Epoxide 56 would be derived from 53 (R = H) through a simple sequence involving partial hydrogenation and stereoselective oxidation.

After diol protection of tetrahydropyran 53, the next step involved stereocontrolled introduction of a secondary alcohol adjacent to the tetrahydropyran ring (Scheme [23]). We performed this transformation by a two-step sequence. Partial hydrogenation with Lindlar catalyst and a Sharpless asymmetric dihydroxylation gave diol 57. Selective tosylation of the primary alcohol group and formation of the terminal epoxy group under basic conditions gave the terminal epoxide 56. This product was opened with an allyl Grignard reagent in the presence of copper(I) iodide. Application of the known osmium tetroxide/sodium periodate protocol to cleave the double bonds, followed by Wittig reaction, and reduction with diisobutylaluminum hydride gave the intermediate allylic alcohol 55. Cyclization of 55 by concomitant asymmetric epoxide formation and cleavage gave the anti-tetrahydrofuran 58 as the sole stereoisomer. The terminal epoxide 54, obtained by a similar sequence to that outlined above, was regioselectively opened by using the suitable Grignard reagent in the presence of copper(I) iodide to give the secondary alcohol 59. This acetonide had previously been transformed into (+)-muconin (15),[56] so our approach constituted a new formal synthesis of this natural product.[57]

As mentioned earlier, polycyclic ether natural products are formed by a cascade cyclization of a polyepoxide backbone. Therefore, epoxide-opening cascades have been used as a valuable method for synthesizing this type of compound.[58] The main drawback of using the polyepoxide-opening strategy is lack of control in the activation of the appropriate epoxide.[59] This affects the yield of the cascade process if more than two epoxide groups are involved. Therefore, the development of mild reaction conditions that permit selective activation of a polyepoxide chain is vital for guiding the cascade event in the correct direction.[60]
Because of the mildness of our procedure, we surmised that the use of a Nicholas cation as a suitable trigger might guide the cyclization process in a polyepoxide chain to give a polycyclic ether in a single step. To test our hypothesis regarding the predictability of the cascade cyclization event, we first decided to test it in a system with two epoxide groups on a carbon chain (Scheme [24]). If the process evolved as expected, that is, through exo cyclization, it should generate two of the three tetrahydrofuran rings of teurilene (11) in one step. However, if the cyclization occurred in an endo manner, rings B and C of the thyrsiferol (12) would be formed.

With the concept of bidirectional synthesis in mind, the diepoxide 61 was easily obtained from commercially available cycloocta-1,5-diene (Scheme [25]).[61] Introduction of the tert-butoxycarbonyl protecting group broke the C2 symmetry of compound 61 and generated a well-documented terminating nucleophile for acid-promoted cascades.[62] The primary alcohol was oxidized and homologated through a Wittig reaction to give the α,β-unsaturated aldehyde 62. Hydrogenation generated the saturated aldehyde, which was immediately coupled with (trimethylsilyl)ethyne anion. The resulting compound was treated with octacarbonyldicobalt to give the model molecule 63, primed for cascade cyclization.

With the bisepoxide 63 in hand, we explored various reaction conditions. We found that silica gel promoted the Nicholas reaction to give a mixture of bitetrahydrofurans 64 and 65 in good yields after two steps (Scheme [26]). Basic treatment of bitetrahydrofurans 64 and 65 gave the cyclic carbonates 66 and 67, which conveniently crystallized, permitting X-ray analysis for confirmation of their structures and stereochemistry.

Having established the expediency of our approach to cascade cyclization, we decided to move forward and synthesize teurilene (11). As shown in the retrosynthetic analysis (Scheme [27]), we surmised that 11 might be obtained through a sequence of reactions starting from the cobalt hexacarbonyl complex 68.
By employing the bidirectional strategy, the known diol 69 was synthesized in good yield in four steps (Scheme [28]).[63] The diol 69 was monoprotected as a silyl ether, leaving the bis(homoallylic) alcohol ready for homologation. By using the protocol developed by our group, we obtained the α,β-unsaturated ester 70 in one step and excellent yield.[64] The ester group was reduced with aluminum hydride to give the corresponding allylic alcohol. The first chiral epoxide was generated by a Katsuki–Sharpless asymmetric epoxidation, and subsequent protection gave compound 71. The silyl group was removed under standard reaction conditions and the resulting alcohol was oxidized to the corresponding aldehyde, which was coupled with lithium (trimethylsilyl)acetylide to give the propargylic alcohol 72. Finally, the remaining epoxides were installed in one step by a Shi asymmetric epoxidation[65] to give the polyepoxide 74.


The formation of the hexacarbonyl dicobalt complex from 74 proceeded smoothly to give the intermediate 68, which we submitted to the cyclization conditions (Scheme [29]). As expected, the cyclization process provided the three tetrahydrofuran rings in excellent yield (75% after two steps). The cascade is high yielding, averaging 91% per epoxide. Although the cascade proceeded efficiently the reaction gave a 1:1 epimeric mixture of 75 and 76 at the propargylic position. From a synthetic standpoint, the epimeric mixture does not represent a problem for the synthesis of 11, because the propargylic ether can be isomerized at a later stage in the synthetic route.

The isomers 75 and 76 were treated under basic conditions to remove the trimethylsilyl group from the alkyne and the tert-butoxycarbonyl group from the primary alcohol. Hydration of the terminal alkyne then gave the methyl ketones 77 and 78. The undesired isomer 78 was converted into the desired isomer 77 by treatment with a base. Transformation of the carbonyl group into the corresponding alkene was achieved through a Wittig reaction. The Cs symmetry of the target molecule was produced by a Sharpless asymmetric dihydroxylation with the AD-mix β, which gave the meso-tetraol 79 exclusively. The meso-tetraol 79 was bidirectionally converted into the diepoxide 80 in good yield by a two-step protocol. Finally, diepoxide 80 was cleaved with a Grignard reagent in the presence of copper(I) iodide to give the natural product teurilene (11) in excellent yield.[66]
3
Ring-Closing Metathesis of Unsaturated Linear Ethers
We were able to prepare six- to nine-membered cyclic ethers (Scheme [19]; R1 = R2 = R3 = H) by means of the intramolecular Nicholas reaction with an alcohol as a nucleophile (the C–O bond-formation strategy).[67] Also, by using this methodology, we were able to obtain an oxane–oxepane system (Scheme [19]; R1–R2 = CH2CH2CH2O; R3 = H, OBn; n = 2). However, this methodology gave poor conversions and poor yields of isolated eight-membered cyclic ethers (n = 3) when R1 ≠ H or R3 ≠ H. Moreover, to obtain fused eight-membered cyclic ethers, such as the oxane–oxocane system (R1– R2 = CH2CH2CH2O; R3 = OBn; n = 3), we needed at least two geometric control elements: a ring and a Z-double bond in the linear chain.[68]
Taking into account this limitation, we considered the possibility of developing a new strategy based on an intermolecular Nicholas reaction (Scheme [30]). Because the RCM reaction has become the most powerful method for preparing cyclic products, we surmised that the cyclic ethers might be formed through a tandem intermolecular Nicholas reaction/RCM (the C–C bond-formation strategy). There are two possible approaches to this tandem strategy; in the first, the alkyne hexacarbonyldicobalt-complexed moiety is located in an endo position with respect to the cyclic ether (path a), and in the second, it is located in an exo position (path b).

By applying the endo strategy (Scheme [30]; path a), we were able to obtain the saturated ethers (+)-cis- lauthisan and (+)-cis-obtusan.[69] Their structures represent the basic skeletons present in a number of naturally occurring nonterpenoidal eight- and nine-membered ring ethers (Scheme [31]). The synthesis of the cobalt-complexed ethers 86 and 87 began with the cobalt complex of commercially available racemic pent-1-yn-3-ol. Intermolecular Nicholas reaction with suitable allylic and homoallylic alcohols gave the α,α′-disubstituted linear ethers 82 and 83, respectively, with almost no stereocontrol of the newly created stereocenter. With ethers 82 and 83 in hand, the preparation of the necessary dienes for the RCM step required only simple alkylation with allyl bromide. Because cyclization to form a medium-sized ring is impractical with a free alkyne as part of the ring, the presence of a cobalt metallic core in 84 and 85 was crucial for ring closure. Compounds 84 and 85 were therefore treated with octacarbonyldicobalt to give the cobalt complexes 86 and 87, respectively.
There are three major advantages to the use of the cobalt alkyne complex in the RCM process. First, the presence of the cobalt complex should prevent the undesirable participation of the triple bond in the metathesis process.[70] Secondly, bending[71] of the acetylenic system as a result of the formation of the cobalt complex might overcome the unfavorable entropic and enthalpic factors associated with the formation of eight- and nine-membered rings (the final ring has an endocyclic triple bond). Thirdly, the hexacarbonyldicobalt–alkyne complex in the endo position can serve as a stereochemical control for a critical isomerization process in the final cyclic product.
RCM was performed by using the second-generation Grubbs catalyst under conditions of high dilution (0.001 M) in refluxing dichloromethane to give the ethers 86 and 87 in excellent yields. Interestingly, at this point of the synthesis the two diastereoisomers of 86 and of 87 could be readily separated by column chromatography on silica gel. However, because the major isomers isolated were trans-86 and trans-87, we considered the possibility of performing isomerizations to give the cis-isomers, because these are usually more stable thermodynamically. Fortunately, the use of montmorillonite K-10 as an acid in the Nicholas reaction proved to be highly efficient in inducing the desired conversion.[72] Thus, the original mixture evolved quantitatively to pleasing cis–trans ratios of 17:1 for the eight-membered ring 88 and 21:1 for the nine-membered ring 89. Reductive decomplexation efficiently gave the corresponding cyclodienes, although these were obtained as inseparable mixtures of stereoisomers at the newly created double bond. However, hydrogenation under standard conditions cleanly gave (+)-cis-lauthisan (90) and (+)-cis-obtusan (91), respectively (Scheme [31]).

However, we could not obtain seven-membered oxacycles by this endo strategy. We therefore developed a powerful and highly convergent method based on the alternative exo approach that permits a very short synthesis of seven-, eight-, or nine-membered cyclic ethers by a genuine tandem intermolecular Nicholas reaction/RCM (Scheme [32]).[73] Our study began with an exploration to determine a suitable position for the double bond in the R2 group of the cobalt complex that would avoid undesirable intramolecular Nicholas reactions or elimination reactions. After some experimentation, we found that complex 92 was the most appropriate for carrying out the intermolecular Nicholas reactions. With allylic, homoallylic, or bishomoallylic alcohols as nucleophiles, this complex gave the corresponding doubly branched ethers in very good yields.


With the diene complexes 93, 94, and 95 in hand, we performed RCM reactions with the second-generation Grubbs catalyst (Scheme [32]). Treatment of the diene cobalt-complexes with 30 mol% of the catalyst in refluxing dichloromethane (0.001 M) cleanly gave the desired cyclic complexes 96 (cis/trans = 1.0:1.1), 97 (cis/trans = 1.1:1.0), and 98, respectively, in excellent yields. When montmorillonite K-10 was used as the acid, the original mixture evolved quantitatively to a cis/trans ratio of 3.0:1.0 for the seven-membered ring 96, and a cis/trans ratio of 5.7:1.0 for the eight-membered ring 97 (Scheme [33]).
The last steps required to complete the synthesis of the basic skeletons present in a number of naturally occurring nonterpenoids were decomplexation of the hexacarbonyldicobalt complexes cis-96 and cis-97, cleavage of the trimethylsilyl group with tetrabutylammonium fluoride, and further catalytic hydrogenation to give the saturated oxacycles cis-101 and (+)-cis-lauthisan (90) in excellent yields (Scheme [33]).
Additionally, by using this strategy, we developed a formal synthesis of the enantiomer of the natural product (+)-laurencin (1).[73] This compound is one of the most representative members of the group of lauroxanes, and was first isolated from the extracts of the red algae L. glandulifera by Irie and co-workers in 1965.[74] Our route to oxocanes seemed well suited for use in conjunction with a total synthesis of (+)-laurencin (1) developed by the Overman group.[75] In this synthesis, the oxocene 102 is an advanced intermediate that is converted in eight steps into the enantiomerically pure (+)-laurencin (1) (Scheme [34]). Synthesis of oxocene 102 would, therefore, represent a formal synthesis of the natural product. As shown in the retrosynthetic analysis in Scheme [34], we surmised that oxocene 102 might be obtained through a simple reactions sequence from the cobalt hexacarbonyl complex cis-103. Alkyne complex cis-103 should be accessible by the three key steps used in our methodology: isomerization of the mixture of diastereomers 103 mediated by montmorillonite K-10 to obtain mainly the cis-isomer, RCM of the unsaturated branched ether cobalt complex 104, and intermolecular Nicholas reaction with the alcohol 105 and the acetylenic cobalt complex 106. However, we were aware that the cobalt complex 106 was not an ideal substrate for the synthesis of cyclic ethers, because the intermolecular Nicholas reaction needed to be carried at –20 ºC to avoid the formation of an elimination product.

After several attempts, we found that the intermolecular Nicholas reaction of cobalt complex 106 with alcohols 105 carrying various protective groups failed, giving mainly the elimination product of the alcohol. Having been unable to construct the desired linear ether, we turned our attention to the design of a new cobalt complex as an electrophile for the Nicholas reaction. This cobalt complex needed to meet two key requirements: first, it had to have a lower tendency to undergo elimination reactions, and secondly, it needed to have a suitable terminal functional group that could be transformed into a double bond in the later stages of the reaction. We found that cobalt complex 107 met these requirements (Scheme [35]). This complex was easily prepared from commercially available ethyl 4-bromobutanoate. Ethyl 4-bromobutanoate was reduced to the corresponding aldehyde, which was treated with lithiated (trimethylsilyl)acetylene to give the propargylic alcohol 108. Complexation of 108 with octacarbonyldicobalt gave the required complex 107.
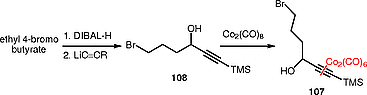
Gratifyingly, when we applied the intermolecular Nicholas reaction to complex 107 using enol 105 (P = TBDPS) as the incoming alcohol, we obtained the corresponding ethers 109 (Scheme [36]). Dienes 104 were readily elaborated from ethers 109 by using a variant of the Grieco reaction.[76] Before the cyclization step by RCM, the alkyne group was complexed with octacarbonyldicobalt to give the cobalt complexes 104 (P = TBDPS). Exposure of dienes 104 (P = TBDPS) to the RCM conditions in the presence of the second-generation Grubbs catalyst gave the eight-membered ring ethers 103 in 99% yields. The oxocenes 103 (P = TBDPS) were obtained as a mixture of cis and trans diastereomers at the propargyl carbon (~1.0:1.0). This mixture of cis and trans isomers, was transformed into the cis-isomer quantitatively by treatment with montmorillonite K-10.

Completion of the formal synthesis from the alkyne–cobalt complex cis-103 (P = TBDPS) involved transformation of the terminal alkyne to an analogue of Overman’s allylic alcohol 111 (Scheme [36]). Although compound 111 has a similar protecting group to that in Overman’s intermediate, and could, therefore, be used to complete the synthesis of the natural product, we decided to check the structure of this compound and to achieve a formal synthesis of (+)-laurencin by changing the protecting group on the secondary alcohol give intermediate 102.
4
Prins Cyclization Promoted by Iron(III) salts
4.1General Strategy
This approach is based on a rapid and direct access to oxygenated ring systems through a Prins cyclization using iron(III) salts as promoters or catalysts. This methodology combines the formation of C–O and C–C bonds in an oxacycle ring in a single step and is complementary to the other methods described above. The cyclization is driven through an oxocarbenium ion intermediate that is generated directly from the corresponding unsaturated alcohol (Scheme [37]).

Previously, this cyclization had been promoted by using a strong acid in a stoichiometric amount with a mixed acetal as the starting material. The main strategy was based on the generation of a mixed acetal (α-acetoxy ether) from the corresponding homoallylic alcohol.[77]
Taking these precedent into accounts, we considered the possibility of directly coupling an unsaturated alcohol with an aldehyde with an iron(III) salt as promoter. This strategy would have the advantages of using a homoallylic or homopropargylic alcohol as the starting material, thereby avoiding the need to generate the mixed acetal, and of using an inexpensive, environmentally friendly, and stable Lewis acid.
4.2
Cyclization Using Alkynols and the Formation of Vinyl Halides
First, to check the catalytic behavior of iron(III) salts in the Prins cyclization, we examined the reaction of but-3-en-1-ol (112) with several aldehydes in the presence of iron(III) chloride or bromide as promoter. The cyclization proceeded satisfactorily with both Lewis acids, giving the corresponding cis-2-alkyl-4-halotetrahydropyrans 113 in good yields (Scheme [38]). We then extended our studies to the alkyne Prins cyclization. In this case, we obtained the corresponding 2-alkyl-4-halo-5,6-dihydro-2H-pyrans 115 in good yields by using but-3-yn-1-ol (114) instead of but-3-en-1-ol (112).[78]

However, when pent-3-yn-1-ol was used, the corresponding tetrahydrofurans 117 (R1 = Me) were the major cyclic products from acetaldehyde (R2 = Me; tetrahydrofuran/tetrahydropyran 35:65; 80%). The structure of the alkyne therefore determines whether endo cyclization to a tetrahydropyran 116 or exo cyclization to a tetrahydrofuran 117 occurs.

In this alkyne Prins cyclization, we found that an unexpected halide exchange occurs with halogenated solvents (Scheme [40]). Through density functional theory studies, we detected an interaction between a halogenated solvent (dibromomethane) and iron(III) chloride that leads to the formation of a complex with a weakened C–Br bond. The presence of the reactive vinyl cation promotes bromine abstraction from the dibromomethane. In the case of the secondary carbocation intermediate of the alkene Prins cyclization, the vinyl cation is insufficiently reactive to promote abstraction of the bromine, leading cleanly to the chlorotetrahydropyran as the sole product. From the synthetic point of view, it is clear that for each halide the corresponding halogenated solvent must be used.[79]

The alkyne Prins cyclization leads to tetrahydropyrans with a halovinyl functionality that can be further derivatized. When we submitted the halovinyl tetrahydropyrans 116 to osmium-catalyzed cis-hydroxylation [Scheme [41](a)], we obtained the corresponding trans-2-alkyl-3-hydroxytetrahydropyran-4-ones stereoselectively as single stereoisomers. This cis-hydroxylation was improved by using a ruthenium methodology, which increased the yield and reduced the reaction time from days to a few minutes [Scheme [41](b)].[80]

Next, we tried to extend this methodology to the synthesis of 2,6-disubstituted dihydropyrans by using a secondary homopropargylic alcohol (Scheme [42]). However, the treatment of pent-4-yn-2-ol with 3-methylbutanal in the presence of iron(III) chloride gave the unsaturated (E)-β-hydroxy ketone 119 and the (E)-α,β-unsaturated ketone 120 in 65% yield, without any trace of the expected Prins-type dihydropyran product.[81]
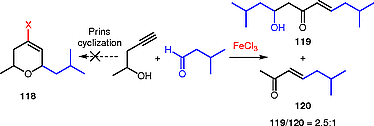
The addition of a secondary homopropargylic alcohol to an aldehyde promoted by an iron(III) halide generates the oxocarbenium ion 121, which can evolve to the Prins cyclic product 118 or undergo a 2-oxonia-[3,3]-sigmatropic rearrangement to give allenolate 122; the latter evolves to the unsaturated linear products 119 and 120 (Scheme [43]).[81] The course of the reaction (rearrangement or alkyne Prins cyclization) depends directly on the stability of the species that are involved in this rearrangement.

4.3
The Use of Trimethylsilyl and Electron-Withdrawing Substituents in the Synthesis of 2,6-Disubstituted Tetrahydropyrans
To suppress the 2-oxonia-[3,3]-sigmatropic rearrangement in favor of the Prins reaction (Scheme [44]), we considered three possible approaches: (a) modification of the bulkiness of the substituent in the aldehyde (R3); (b) the introduction of a trimethylsilyl group onto the terminal carbon of the alkyne, based on the well-documented silyl-modified Prins cyclization; and (c) destabilization of the allenic cation.

When the bulkiness of R3 was increased from methyl to cyclohexyl, Prins dihydropyrans 118 were obtained as the major products. However, this is a particular case that does not permit full control over the balance between sigmatropic rearrangement and alkyne Prins cyclization. In contrast, the presence of a trimethylsilyl group on the alkyne moiety favored the cyclization and suppressed the rearrangement as a competing pathway.
When we carried out a silylalkyne Prins cyclization by reacting a secondary trimethylsilyl homopropargylic alcohol 123 with an aldehyde in the presence of iron(III) chloride or bromide as a promoter (Scheme [44]), we obtained the six-membered ring product exclusively and in good yield. No traces of products resulting from the oxonia rearrangement were observed, and we were able to prepare tetra- and pentasubstituted dihydropyrans in one step by using suitable unsaturated alcohol.[82] The presence of the silyl group on the alkyne moiety is essential for the reaction; when the alkyne is substituted with a methyl group, the process is inhibited. Furthermore, the size of the substituents on the silicon atom is also a critical factor. For instance, when the alkyne moiety is substituted by a triisopropylsilyl group instead of trimethylsilyl group, cyclization does not take place.
Surprisingly, the resulting halosilyl alkenes 124 proved to be very unreactive systems. For instance, all attempts to cleave the C–Si bond under standard conditions were unsuccessful. Fortunately, however, the trimethylsilyl group was satisfactorily cleaved by refluxing the silylated alkene in aqueous hydriodic acid for one week (Scheme [45]). Despite the long reaction time, the corresponding desilylated dihydropyrans 125 were obtained in excellent yields with complete retention of the cis configuration.

Because the incorporation of a trimethylsilyl group as a substituent in homopropargylic alcohols 123 favors their Prins cyclization by destabilizing the allenic cation 122, we reasoned that similar results might be obtained by introducing other types of substituent that destabilize the corresponding allenic cation and, consequently, accelerate the Prins cyclization.[83] We decided to study the effects of electron-withdrawing groups, such as chloro or nitrile groups located in the position α to the hydroxy group, on the relative rates of ring opening versus alkyne Prins cyclization. With chloromethyl or cyanomethyl substituents (R1 = CH2Cl or CH2CN) as electron-withdrawing groups and a bulky aldehyde, such as cyclohexanecarbaldehyde (R3 = Cy), the corresponding cis-2,6-disubstituted dihydropyrans 125 (R2 = H) were the main products (Scheme [46]).[84]

The relative stability of intermediate species involved in 2-oxonia-[3,3]-sigmatropic rearrangement (Scheme [43]) is the key factor to consider, because this can favor the Prins cyclization or the corresponding domino process. Therefore, we performed density functional theory studies on simplified structures. The results of these calculations are summarized in the energy diagrams shown in Figure [2].

The α-silane allenyl cation 128 is 5.7 kcal/mol less stable than the homopropargylic analogue 126, which indicates that the rearrangement is not favored, and the dihydropyran 127 is protected from ring opening by an activation energy of 1.7 kcal/mol. The rearrangement is an endothermic process, and is less favored than the reaction without silicon (133 to 132). Furthermore, replacement of the silane substituent by a trimethylsilyl group results in an even less favorable rearrangement of 129 to 131. This energy profile correlates very well with our proposed mechanism.

Density functional theory calculations suggested that the trimethylsilyl group stabilizes the dihydropyranyl cations in the dihydropyranylium cation 130, leading to more-uniform reorganization of the electronic density in the ring. The calculations also showed that the trimethylsilyl group does not have a direct effect on the formally positively charged carbon atom.[84]
To confirm that no racemization occurred during the cyclizations to the tetrasubstituted dihydropyrans, we treated alcohol (R)-134 with phenylacetaldehyde and iron(III) chloride. The resulting dihydropyran 135 showed a slightly reduced but still synthetically useful enantiomeric purity compared with the starting homopropargylic alcohol (R)-134 (Scheme [48]).

4.4
Catalytic Prins Cyclization
Next, with the aim of achieving greater sustainability, we focused on the development of a catalytic Prins cyclization (Scheme [49]). We made considerable efforts to replace stoichiometric methods by catalytic processes, the halide source being our first concern. Trimethylsilyl halides were chosen, based on precedents with indium salts.[85] Catalytic systems obtained by combining readily available iron salts with trimethylsilyl halides are widely applicable and promote the formation of substituted six-membered oxacycles.[86] The key finding for this protocol is that a catalytic effect is produced by a combination of a readily available and inexpensive iron source [FeX3 or Fe(acac)3] and a trimethylsilyl halide. This catalytic method also permits the construction of chloro, bromo, or iodo heterocycles by appropriate selection of the iron(III) source, the trimethylsilyl halide, and the solvent.

A tentative catalytic cycle is shown in Scheme [50]. We reasoned that the catalytic cycle might be initiated through activation of the aldehyde by the iron salt. The activated aldehyde then reacts with the unsaturated nucleophilic species to give an acetal precursor. The high stability of the accumulating iron oxide counterpart and the fact that the iron salt is the sole source of halide provide a thermodynamic sink that drives the conversion, but requires the use of a stoichiometric or excess amount of the iron(III) salt. We therefore devised an indirect way to complete the catalytic cycle through ligand exchange between the iron complex and a chlorosilane to regenerate the iron(III) halide as a result of the more oxyphilic character of the silicon.

4.5
Synthesis of Oxepanes
Having gained experience with the Prins cyclization described above, we focused our attention on developing a direct synthesis of oxepanes, for which a few precedents are found in the literature.[87] [88] Our studies on the direct synthesis of oxepanes began with the Prins cyclization of pent-4-en-1-ol (136, R1 = H) with 3-methylbutanal in dry dichloromethane in the presence of a stoichiometric amount of iron(III) chloride (Scheme [51]). The desired all cis-disubstituted oxepane 137 was obtained in 56% yield, together with 41% of the monosubstituted tetrahydrofuran 138 (Scheme [51]).[89] By using more-hindered aldehydes (R2= Cy, t-Bu), the corresponding oxepane 137 was obtained exclusively, with none of the corresponding tetrahydrofuran. When we tested the cyclization of secondary bishomoallylic alcohols (136, R1 ≠ H) under the same conditions, we obtained the all-cis 2,4,7-trisubstituted oxepanes 137 as the major products. Again, with a hindered aldehyde such as pivalaldehyde, the corresponding oxepane 137 was formed exclusively in good yield (75%).[90]

The presence of the tetrahydrofuran derivatives 138 prompted us to use our previously described catalytic system of an iron(III) salt and trimethylsilyl chloride. The application of this catalytic system to the Prins cyclization gave oxepanes in good yields, with none of the corresponding tetrahydrofuran derivative 138 (Scheme [52]). With bishomoallylic alcohols 136, iron(III) chloride or tris(acetylacetonate)iron catalyzed the cyclization to give, in both cases, a mixture of cis-2,7-dialkyloxepanes 137 and 139, differing in the stereochemistry of the chlorine atom at C-4. The use of tris(acetylacetonate)iron produced better results with aldehydes bearing functional groups such as double bonds. It should be emphasized that the excellent yields obtained from functionalized aldehydes with both iron(III) sources provides a route to other functional groups. The Prins cyclization generally led to 60:40 mixtures of oxepanes 137 and 139, respectively, except when a bulky aldehyde was used. From a synthetic point of view, the absence of stereochemical control at the chlorine-substituted carbon is not a limitation for the effective synthesis of the oxepane ring, as the chlorine atom can be easily removed to give a single cis-2,7-disubstituted oxepane.

On the basis of our experimental observations, we proposed a mechanism involving the catalytic formation of the oxocarbenium ion 140. The tetrahydrofuran 138 might be generated from the bicycle 143 (Scheme [53]). In the oxepane 139, the ether oxygen participates in a transannular cyclization by an SN2 mechanism as a result of the optimal spatial requirement for the chlorine atom (internal O–C–Cl angle 166.7º), leading to the bicycle 143. The attack of the chloro group on the carbon adjacent to the oxonium, with R2 as substituent, leads to the thermodynamically more stable tetrahydrofuran 138. In the case of bulky substituents at this position (R2 = Cy, t-Bu), this attack is unfavorable, and the presence of tetrahydrofuran 138 is not detected. However, in the oxepane 137, the transannular cyclization is unfavorable, because the oxygen and chlorine (internal O–C–Cl angle 141.7º) does not meet the spatial requirement for formation of the bicycle 141 and, therefore, the tetrahydrofuran 142.

To validate our methodology, we concentrated our efforts on a total synthesis of (+)-isolaurepan with a Prins cyclization as the key step (Scheme [54]). We planned this synthesis from the enantiomerically enriched bishomoallylic alcohol 145, containing a hexyl group, which was obtained in one step. Regioselective ring opening of the known epoxide (–)-144 with allylmagnesium bromide in the presence of copper(I) iodide gave the desired bishomoallylic alcohol 145 in 90% yield (>99 ee). Prins cyclization of 145 with butanal was performed under standard reaction conditions with tris(acetylacetonate)iron as the iron(III) source to give the desired oxepane 146 in 95% yield. A final dehalogenation step gave (+)-isolaurepan in 90% yield. Starting from the enantiomerically enriched epoxide 144, we accomplished the most efficient and the shortest reported total synthesis of (+)-isolaurepan with 77% overall yield.
5
Summary
Cyclic ether moieties are very important structural motifs in many biologically active natural products. In this account, we have described several approaches that our group has adopted in synthesizing polysubstituted cyclic ethers. We have paid a great deal of attention to the biogenesis of many of these compounds as a source of inspiration for planning of their syntheses. Three main approaches have been adopted. These are based, respectively, on the formation of a C–O bond as the key step, the construction of the cyclic ether by an RCM reaction in a preconstructed linear ether, and the synthesis of the oxygenated heterocycle by concomitant C–O and C–C bond formation in a Prins cyclization.

Acknowledgment
This research was supported by the Spanish MINECO, co-financed by the European Regional Development Fund (ERDF) (CTQ2011-28417-C02-01/BQU and CTQ2011-22653) and the IMBRAIN project (FP7-REGPOT-2012-CT2012-31637-IMBRAIN), which is funded under the Seventh Framework Programme (CAPACITIES). We acknowledge our co-workers who participated in this research program, and whose names appear in the reference section. The authors thank Dr. David Tejedor for helpful discussions.
-
References
- 1 Rutkowski J, Brzezinski B. Biomed. Res. Int. 2013; 2013: 162513
- 2 Kadota I, Yamamoto Y. Acc. Chem. Res. 2005; 38: 423
- 3 Li N, Shi Z, Tang Y, Chen J, Li X. Beilstein J. Org. Chem. 2008; 4: doi: 10.3762/bjoc.4.48
- 4 Wang B.-G, Gloer JB, Ji N.-Y, Zhao J.-C. Chem. Rev. 2013; 113: 3632
- 5a Nakata T. Chem. Rev. 2005; 105: 4314
- 5b Nakata T. Chem. Soc. Rev. 2010; 39: 1955
- 5c Inoue M. Chem. Rev. 2005; 105: 4379
- 5d Kadota I, Yamamoto Y. Acc. Chem. Res. 2005; 38: 423
- 5e Vilotijevic I, Jamison TF. Angew. Chem. Int. Ed. 2009; 48: 5250
- 5f Deiters A, Martin SF. Chem. Rev. 2004; 104: 2199
- 5g Alvarez E, Candenas ML, Pérez R, Ravelo JL, Martín JD. Chem. Rev. 1995; 95: 1953
- 5h Morris JC, Phillips AJ. Nat. Prod. Rep. 2011; 28: 269 ; and previous issues in the series
- 6 Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. Angew. Chem. Int. Ed. 2000; 39: 44
- 7a Nicolaou KC In Classics in Total Synthesis: Targets, Strategies, Methods . Nicolaou KC, Sorensen EJ. VCH; Weinheim: 1996. Chap. 37, 731
- 7b Hansen MC, Verdaguer X, Buchwald SL. J. Org. Chem. 1998; 63: 2360
- 7c Schreiber SL, Kelly SE, Porco JA, Sammakia T, Suh EM. J. Am. Chem. Soc. 1988; 110: 6210
- 7d Burton JW, Clark JS, Derrer S, Stork TC, Bendall JG, Holmes AB. J. Am. Chem. Soc. 1997; 119: 7483
- 8a Crimmins MT, Ellis JM. J. Am. Chem. Soc. 2005; 127: 17200
- 8b Hirama M, Oishi T, Uehara H, Inoue M, Maruyama M, Guri H, Satake M. Science 2001; 294: 1904
- 9a Crane EA, Scheidt KA. Angew. Chem. Int. Ed. 2010; 49: 8316
- 9b Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron 2010; 66: 413
- 9c Pastor IM, Yus M. Curr. Org. Chem. 2007; 11: 925
- 10a Moore RE. In. Marine Natural Products . Vol. 1. Scheuer PJ. Academic Press; New York: 1978: 43
- 10b Erickson KL In Marine Natural Products . Vol. 5. Scheuer PJ. Academic Press; New York: 1983: 131
- 10c Faulkner DJ. Nat. Prod. Rep. 2002; 19: 1 and previous issues in the series
- 10d Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Nat. Prod. Rep. 2013; 30: 237 ; and previous issues in the series
- 11 Kurosawa E, Fukuzawa A, Irie T. Tetrahedron Lett. 1972; 21: 2121
- 12 Suzuki T, Koizumi K, Suzuki M, Kurosawa E. Chem. Lett. 1983; 1643
- 13a Fukuzawa A, Aye M, Takasugi Y, Nakamura M, Tamura M, Murai A. Chem. Lett. 1994; 2307
- 13b Fukuzawa A, Aye M, Murai A. Chem. Lett. 1990; 1579
- 14 Warren RG, Wells RJ, Blount JF. Aust. J. Chem. 1980; 33: 891
- 15 Capon RJ, Barrow RA, Rochfort S, Jobling M, Skene C. Tetrahedron 1998; 54: 2227
- 16a Capon RJ, Barrow RA, Skene C, Rochfort S. Tetrahedron Lett. 1997; 38: 7609
- 16b Capon RJ, Barrow RA. J. Org. Chem. 1998; 63: 75
- 17a Fernández JJ, Souto ML, Norte M. Nat. Prod. Rep. 2000; 17: 235
- 17b Pacheco FC, Villa-Pulgarin JA, Mollinedo F, Martín MN, Fernández JJ, Daranas AH. Mar. Drugs 2011; 9: 2220
- 17c Cen-Pacheco F, Rodríguez J, Norte M, Fernández JJ, Hernández Daranas A. Chem. Eur. J. 2013; 19: 8525
- 18 Suzuki T, Suzuki M, Furusaki A, Matsumoto T, Kato A, Imanaka Y, Kurosawa E. Tetrahedron Lett. 1985; 26: 1329
- 19 Blunt JW, Hartshorn MP, McLeannan TJ, Munro MH. G, Robinson WT, Yorke SC. Tetrahedron Lett. 1978; 19: 69
- 20 Satake M, Murata M, Yasumoto T. Tetrahedron Lett. 1993; 34: 1975
- 21a Shimizu Y, Chou H.-N, Bando H, Van Duyne G, Clardy J. J. Am. Chem. Soc. 1986; 108: 514
- 21b Shimizu Y, Bando H, Chou H.-N, Van Duyne G, Clardy J. J. Chem. Soc., Chem. Commun. 1986; 1656
- 22 Lin YY, Risk M, Ray SM, Van Engen D, Clardy J, Golik J, James JC, Nakanishi K. J. Am. Chem. Soc. 1981; 103: 6773
- 23 Murata M, Iwashita T, Yokoyama A, Sasaki M, Yasumoto T. J. Am. Chem. Soc. 1992; 114: 6594
- 24a Murata M, Kumagai M, Lee JS, Yasumoto T. Tetrahedron Lett. 1987; 28: 5869
- 24b Takahashi H, Kusumi T, Kan Y, Satake M, Yasumoto T. Tetrahedron Lett. 1996; 37: 7087
- 25 Nakanishi K. Toxicon 1985; 23: 473
- 26 Bermejo A, Figadère B, Zafra-Polo M.-C, Barrachina I, Estornell E, Cortes D. Nat. Prod. Rep. 2005; 22: 269
- 27a Alali FQ, Liu XX, McLaughlin JL. J. Nat. Prod. 1999; 62: 504
- 27b Zeng L, Ye Q, Oberlies NH, Shi G, Gu Z.-M, He K, McLaughlin JL. Nat. Prod. Rep. 1996; 13: 275
- 28a Añorbe B, Martín VS, Palazón JM, Trujillo JM. Tetrahedron Lett. 1986; 27: 4991
- 28b Palazón JM, Martín VS. Tetrahedron 1988; 29: 681
- 28c Tonn CE, Palazón JM, Ruíz-Pérez C, Rodríguez ML, Martín VS. Tetrahedron Lett. 1988; 29: 3149
- 28d Martín VS, Palazón JM. Tetrahedron Lett. 1992; 33: 2399
- 29 Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994; 94: 2483
- 30 Ragoussis N. Tetrahedron Lett. 1987; 28: 93
- 31 Katsuki T, Martín VS. Org. React. (N. Y.) 1996; 48: 1
- 32 Comprehensive Asymmetric Catalysis . Vol. II. Jacobsen EN, Pfaltz A, Yamamoto H. Springer; Berlin: 1999. and references cited therein
- 33 García C, Soler MA, Martín VS. Tetrahedron Lett. 2000; 41: 4127
- 34 Boukouvalas J, Fortier G, Radu I.-I. J. Org. Chem. 1998; 63: 916
- 35 Garcia C, Martín T, Martín VS. J. Org. Chem. 2001; 66: 1420
- 36 Martín T, Martín VS. Tetrahedron Lett. 2000; 41: 2503
- 37 Martín, T.; Martín, V. S., unpublished results.
- 38 Taber DF, You K. J. Org. Chem. 1995; 60: 139
- 39 Yamaguchi M, Hirao I. J. Chem. Soc., Chem. Commun. 1984; 202
- 40 Corey EJ, Ruden RA. Tetrahedron Lett. 1973; 14: 1495
- 41 Martín T, Soler MA, Betancort JM, Martín VS. J. Org. Chem. 1997; 62: 1570
- 42 Baldwin JE. J. Chem. Soc., Chem. Commun. 1976; 734
- 43a Nicolaou KC, Prasad CV. C, Somers PK, Hwang CK. J. Am. Chem. Soc. 1989; 111: 5330
- 43b Mori Y, Yaegashi K, Furukawa H. J. Am. Chem. Soc. 1996; 118: 8158
- 43c Tokiwano T, Fujiwara K, Murai A. Synlett 2000; 335
- 43d Simpson GL, Heffron TP, Merino E, Jamison TF. J. Am. Chem. Soc. 2006; 128: 1056
- 43e Tong R, Valentine JC, McDonald FE, Cao R, Fang X, Hardcastle KI. J. Am. Chem. Soc. 2007; 129: 1050
- 43f Wan S, Gunaydin H, Houk KN, Floreancig PE. J. Am. Chem. Soc. 2007; 129: 7915
- 44 Vilotijevic I, Jamison TF. Science 2007; 317: 1189
- 45 Rawlings BJ, Reese PB, Ramer SE, Vederas JC. J. Am. Chem. Soc. 1989; 111: 3382
- 46a Martín VS, Nuñez MT, Ramírez MA, Soler MA. Tetrahedron Lett. 1990; 31: 763
- 46b Palazón JM, Soler MA, Ramírez MA, Martín VS. Tetrahedron Lett. 1993; 34: 5467 ; see also ref. 28d
- 47 Martín VS, Nuñez T, Tonn CE. Tetrahedron Lett. 1988; 29: 2701
- 48 Ando K. Tetrahedron Lett. 1995; 36: 4105
- 49 Ramírez MA, Padrón JM, Palazón JM, Martín VS. J. Org. Chem. 1997; 62: 4584
- 50 Hoffmann RW. Angew. Chem. Int. Ed. Engl. 1992; 31: 1124
- 51 Betancort JM, Martín VS, Padrón JM, Palazón JM, Ramírez MA, Soler MA. J. Org. Chem. 1997; 62: 4570
- 52 Soler MA, Palazón JM, Martín VS. Tetrahedron Lett. 1993; 34: 5471
- 53 Nicholas KM. Acc. Chem. Res. 1987; 20: 207
- 54 For early synthetic applications of the Nicholas reaction from our group, see: Díaz DD, Betancort JM, Martín VS. Synlett 2007; 343
- 55 Crisóstomo FR. P, Martín T, Martín VS. Org. Lett. 2004; 6: 565
- 56 Takahashi S, Kubota A, Nakata T. Tetrahedron 2003; 59: 1627
- 57 Crisóstomo FR. P, Carrillo R, León LG, Martín T, Padrón JM, Martín VS. J. Org. Chem. 2006; 71: 2339
- 58a Xiong Z, Corey EJ. J. Am. Chem. Soc. 2000; 122: 9328
- 58b Mi Y, Schreiber JV, Corey EJ. J. Am. Chem. Soc. 2002; 124: 11290
- 59a McDonald FE, Wang X, Do B, Hardcastle KI. Org. Lett. 2000; 2: 2917
- 59b McDonald FE, Bravo F, Wang X, Wei X, Toganoh M, Rodríguez JR, Do B, Neiwert WA, Hardcastle KI. J. Org. Chem. 2002; 67: 2515
- 59c Xiong Z, Busch R, Corey EJ. Org. Lett. 2010; 12: 1512
- 60 Tanuwidjaja J, Ng S.-S, Jamison TF. J. Am. Chem. Soc. 2009; 131: 12084
- 61a Lindel T, Franck B. Tetrahedron Lett. 1995; 36: 9465
- 61b Enders D, Schüßeler T. Synthesis 2002; 2280
- 62 Bravo F, McDonald FE, Neiwert WA, Do B, Hardcastle KI. Org. Lett. 2003; 5: 2123
- 63 Johnson WS, Werthemann L, Bartlett WR, Brocksom TJ, Li T.-T, Faulkner DJ, Petersen MR. J. Am. Chem. Soc. 1970; 92: 741
- 64 Crisóstomo FR. P, Carrillo R, Martín T, García-Tellado F, Martín VS. J. Org. Chem. 2005; 70: 10099
- 65 Wong OA, Shi Y. Chem. Rev. 2008; 108: 3958
- 66 Rodríguez-López J, Crisóstomo FP, Ortega N, López-Rodríguez M, Martín VS, Martín T. Angew. Chem. Int. Ed. 2013; 52: 3659
- 67a Palazón JM, Martín VS. Tetrahedron Lett. 1995; 36: 3549
- 67b Díaz DD, Martín T, Martín VS. Org. Lett. 2001; 3: 3289
- 67c Díaz DD, Betancort JM, Crisóstomo FR. P, Martín T. Tetrahedron 2002; 58: 1913
- 68 Betancort JM, Martín T, Palazón JM, Martín VS. J. Org. Chem. 2003; 68: 3216
- 69a Ortega N, Martín T, Martín VS. Org. Lett. 2006; 8: 871
- 69b Ortega N, Martín T, Martín VS. Eur. J. Org. Chem. 2009; 554
- 70 For representative examples of enyne metathesis, see: Diver ST, Giessert AJ. Chem. Rev. 2004; 104: 1317
- 71a Green JR. Synlett 2001; 353
- 71b Burlison JA, Gray JM, Young DG. J. Tetrahedron Lett. 2001; 42: 5363
- 71c Yang Z.-Q, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. J. Am. Chem. Soc. 2004; 126: 7881
- 72 Crisóstomo FR. P, Carrillo R, Martín T, Martín VS. Tetrahedron Lett. 2005; 46: 2829
- 73 Ortega N, Martín VS, Martín T. J. Org. Chem. 2010; 75: 6660
- 74 Irie T, Suzuki M, Masamune T. Tetrahedron Lett. 1965; 6: 1091
- 75 Bratz M, Bullock WH, Overman LE, Takemoto T. J. Am. Chem. Soc. 1995; 117: 5958
- 76a Sharpless KB, Young MW. J. Org. Chem. 1974; 40: 947
- 76b Burke SD, Voight EA. Org. Lett. 2001; 237
- 77a Marumoto S, Jaber JJ, Vitale JP, Rychnovsky SD. Org. Lett. 2002; 4: 3919
- 77b Pastor IM, Yus M. Curr. Org. Chem. 2007; 11: 925
- 77c Pastor IM, Yus M. Curr. Org. Chem. 2012; 16: 1277
- 78 Miranda PO, Díaz DD, Padrón JI, Bermejo J, Martín VS. Org. Lett. 2003; 5: 1979
- 79 Miranda PO, Carballo RM, Ramírez MA, Martín VS, Padrón JI. ARKIVOC 2007; (iv): 331
- 80 Miranda PO, Padrón JM, Padrón JI, Villar J, Martín VS. ChemMedChem 2006; 1: 323
- 81 Miranda PO, Ramírez MA, Padrón JI, Martín VS. Tetrahedron Lett. 2006; 47: 283
- 82 Miranda PO, Ramírez MA, Padrón JI, Martín VS. Org. Lett. 2006; 8: 1633
- 83a Jasti R, Anderson CD, Rychnovsky SD. J. Am. Chem. Soc. 2005; 127: 9939
- 83b Jasti R, Rychnovsky SD. J. Am. Chem. Soc. 2006; 128: 13640
- 84 Miranda PO, Ramírez MA, Martín VS, Padrón JI. Chem. Eur. J. 2008; 14: 6260
- 85a Onishi Y, Ito T, Yasuda M, Baba A. Eur. J. Org. Chem. 2002; 1578
- 85b Onishi Y, Ito T, Yasuda M, Baba A. Tetrahedron 2002; 58: 8227
- 85c Chan K.-P, Loh T.-P. Org. Lett. 2005; 7: 4491
- 85d Chan K.-P, Seow A.-H, Loh T.-P. Tetrahedron Lett. 2007; 48: 37
- 85e Chan K.-P, Ling YH, Loh T.-P. Chem. Commun. 2007; 939
- 85f Liu F, Loh T.-P. Org. Lett. 2007; 9: 2063
- 86 Miranda PO, Carballo RM, Martín VS, Padrón JI. Org. Lett. 2009; 11: 357
- 87a Coppi L, Ricci A, Taddei M. J. Org. Chem. 1988; 53: 913
- 87b Lolkema LD. M, Hiemstra H, Mooiweer HH, Speckamp WN. Tetrahedron Lett. 1988; 29: 6365
- 87c Furman B, Dziedzic M, Justyniak I. Tetrahedron 2008; 64: 3103
- 88a Overman LE, Castañeda A, Blumenkopf TA. J. Am. Chem. Soc. 1986; 108: 1303
- 88b Castañeda A, Kucera DJ, Overman LE. J. Org. Chem. 1989; 54: 5695
- 88c Berger D, Overman LE, Renhowe PA. J. Am. Chem. Soc. 1997; 119: 2446
- 89 Purino MA, Ramírez MA, Daranas AH, Martín VS, Padrón JI. Org. Lett. 2012; 14: 5904
- 90 The relative stereochemistry of the tetrahydrofurans was determined by means of NMR spectroscopy, see: Napolitano JG, Gavín JA, García C, Norte M, Fernández JJ, Daranas AH. Chem. Eur. J. 2011; 17: 6338
For preliminary results, see:
-
References
- 1 Rutkowski J, Brzezinski B. Biomed. Res. Int. 2013; 2013: 162513
- 2 Kadota I, Yamamoto Y. Acc. Chem. Res. 2005; 38: 423
- 3 Li N, Shi Z, Tang Y, Chen J, Li X. Beilstein J. Org. Chem. 2008; 4: doi: 10.3762/bjoc.4.48
- 4 Wang B.-G, Gloer JB, Ji N.-Y, Zhao J.-C. Chem. Rev. 2013; 113: 3632
- 5a Nakata T. Chem. Rev. 2005; 105: 4314
- 5b Nakata T. Chem. Soc. Rev. 2010; 39: 1955
- 5c Inoue M. Chem. Rev. 2005; 105: 4379
- 5d Kadota I, Yamamoto Y. Acc. Chem. Res. 2005; 38: 423
- 5e Vilotijevic I, Jamison TF. Angew. Chem. Int. Ed. 2009; 48: 5250
- 5f Deiters A, Martin SF. Chem. Rev. 2004; 104: 2199
- 5g Alvarez E, Candenas ML, Pérez R, Ravelo JL, Martín JD. Chem. Rev. 1995; 95: 1953
- 5h Morris JC, Phillips AJ. Nat. Prod. Rep. 2011; 28: 269 ; and previous issues in the series
- 6 Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. Angew. Chem. Int. Ed. 2000; 39: 44
- 7a Nicolaou KC In Classics in Total Synthesis: Targets, Strategies, Methods . Nicolaou KC, Sorensen EJ. VCH; Weinheim: 1996. Chap. 37, 731
- 7b Hansen MC, Verdaguer X, Buchwald SL. J. Org. Chem. 1998; 63: 2360
- 7c Schreiber SL, Kelly SE, Porco JA, Sammakia T, Suh EM. J. Am. Chem. Soc. 1988; 110: 6210
- 7d Burton JW, Clark JS, Derrer S, Stork TC, Bendall JG, Holmes AB. J. Am. Chem. Soc. 1997; 119: 7483
- 8a Crimmins MT, Ellis JM. J. Am. Chem. Soc. 2005; 127: 17200
- 8b Hirama M, Oishi T, Uehara H, Inoue M, Maruyama M, Guri H, Satake M. Science 2001; 294: 1904
- 9a Crane EA, Scheidt KA. Angew. Chem. Int. Ed. 2010; 49: 8316
- 9b Olier C, Kaafarani M, Gastaldi S, Bertrand MP. Tetrahedron 2010; 66: 413
- 9c Pastor IM, Yus M. Curr. Org. Chem. 2007; 11: 925
- 10a Moore RE. In. Marine Natural Products . Vol. 1. Scheuer PJ. Academic Press; New York: 1978: 43
- 10b Erickson KL In Marine Natural Products . Vol. 5. Scheuer PJ. Academic Press; New York: 1983: 131
- 10c Faulkner DJ. Nat. Prod. Rep. 2002; 19: 1 and previous issues in the series
- 10d Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Nat. Prod. Rep. 2013; 30: 237 ; and previous issues in the series
- 11 Kurosawa E, Fukuzawa A, Irie T. Tetrahedron Lett. 1972; 21: 2121
- 12 Suzuki T, Koizumi K, Suzuki M, Kurosawa E. Chem. Lett. 1983; 1643
- 13a Fukuzawa A, Aye M, Takasugi Y, Nakamura M, Tamura M, Murai A. Chem. Lett. 1994; 2307
- 13b Fukuzawa A, Aye M, Murai A. Chem. Lett. 1990; 1579
- 14 Warren RG, Wells RJ, Blount JF. Aust. J. Chem. 1980; 33: 891
- 15 Capon RJ, Barrow RA, Rochfort S, Jobling M, Skene C. Tetrahedron 1998; 54: 2227
- 16a Capon RJ, Barrow RA, Skene C, Rochfort S. Tetrahedron Lett. 1997; 38: 7609
- 16b Capon RJ, Barrow RA. J. Org. Chem. 1998; 63: 75
- 17a Fernández JJ, Souto ML, Norte M. Nat. Prod. Rep. 2000; 17: 235
- 17b Pacheco FC, Villa-Pulgarin JA, Mollinedo F, Martín MN, Fernández JJ, Daranas AH. Mar. Drugs 2011; 9: 2220
- 17c Cen-Pacheco F, Rodríguez J, Norte M, Fernández JJ, Hernández Daranas A. Chem. Eur. J. 2013; 19: 8525
- 18 Suzuki T, Suzuki M, Furusaki A, Matsumoto T, Kato A, Imanaka Y, Kurosawa E. Tetrahedron Lett. 1985; 26: 1329
- 19 Blunt JW, Hartshorn MP, McLeannan TJ, Munro MH. G, Robinson WT, Yorke SC. Tetrahedron Lett. 1978; 19: 69
- 20 Satake M, Murata M, Yasumoto T. Tetrahedron Lett. 1993; 34: 1975
- 21a Shimizu Y, Chou H.-N, Bando H, Van Duyne G, Clardy J. J. Am. Chem. Soc. 1986; 108: 514
- 21b Shimizu Y, Bando H, Chou H.-N, Van Duyne G, Clardy J. J. Chem. Soc., Chem. Commun. 1986; 1656
- 22 Lin YY, Risk M, Ray SM, Van Engen D, Clardy J, Golik J, James JC, Nakanishi K. J. Am. Chem. Soc. 1981; 103: 6773
- 23 Murata M, Iwashita T, Yokoyama A, Sasaki M, Yasumoto T. J. Am. Chem. Soc. 1992; 114: 6594
- 24a Murata M, Kumagai M, Lee JS, Yasumoto T. Tetrahedron Lett. 1987; 28: 5869
- 24b Takahashi H, Kusumi T, Kan Y, Satake M, Yasumoto T. Tetrahedron Lett. 1996; 37: 7087
- 25 Nakanishi K. Toxicon 1985; 23: 473
- 26 Bermejo A, Figadère B, Zafra-Polo M.-C, Barrachina I, Estornell E, Cortes D. Nat. Prod. Rep. 2005; 22: 269
- 27a Alali FQ, Liu XX, McLaughlin JL. J. Nat. Prod. 1999; 62: 504
- 27b Zeng L, Ye Q, Oberlies NH, Shi G, Gu Z.-M, He K, McLaughlin JL. Nat. Prod. Rep. 1996; 13: 275
- 28a Añorbe B, Martín VS, Palazón JM, Trujillo JM. Tetrahedron Lett. 1986; 27: 4991
- 28b Palazón JM, Martín VS. Tetrahedron 1988; 29: 681
- 28c Tonn CE, Palazón JM, Ruíz-Pérez C, Rodríguez ML, Martín VS. Tetrahedron Lett. 1988; 29: 3149
- 28d Martín VS, Palazón JM. Tetrahedron Lett. 1992; 33: 2399
- 29 Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994; 94: 2483
- 30 Ragoussis N. Tetrahedron Lett. 1987; 28: 93
- 31 Katsuki T, Martín VS. Org. React. (N. Y.) 1996; 48: 1
- 32 Comprehensive Asymmetric Catalysis . Vol. II. Jacobsen EN, Pfaltz A, Yamamoto H. Springer; Berlin: 1999. and references cited therein
- 33 García C, Soler MA, Martín VS. Tetrahedron Lett. 2000; 41: 4127
- 34 Boukouvalas J, Fortier G, Radu I.-I. J. Org. Chem. 1998; 63: 916
- 35 Garcia C, Martín T, Martín VS. J. Org. Chem. 2001; 66: 1420
- 36 Martín T, Martín VS. Tetrahedron Lett. 2000; 41: 2503
- 37 Martín, T.; Martín, V. S., unpublished results.
- 38 Taber DF, You K. J. Org. Chem. 1995; 60: 139
- 39 Yamaguchi M, Hirao I. J. Chem. Soc., Chem. Commun. 1984; 202
- 40 Corey EJ, Ruden RA. Tetrahedron Lett. 1973; 14: 1495
- 41 Martín T, Soler MA, Betancort JM, Martín VS. J. Org. Chem. 1997; 62: 1570
- 42 Baldwin JE. J. Chem. Soc., Chem. Commun. 1976; 734
- 43a Nicolaou KC, Prasad CV. C, Somers PK, Hwang CK. J. Am. Chem. Soc. 1989; 111: 5330
- 43b Mori Y, Yaegashi K, Furukawa H. J. Am. Chem. Soc. 1996; 118: 8158
- 43c Tokiwano T, Fujiwara K, Murai A. Synlett 2000; 335
- 43d Simpson GL, Heffron TP, Merino E, Jamison TF. J. Am. Chem. Soc. 2006; 128: 1056
- 43e Tong R, Valentine JC, McDonald FE, Cao R, Fang X, Hardcastle KI. J. Am. Chem. Soc. 2007; 129: 1050
- 43f Wan S, Gunaydin H, Houk KN, Floreancig PE. J. Am. Chem. Soc. 2007; 129: 7915
- 44 Vilotijevic I, Jamison TF. Science 2007; 317: 1189
- 45 Rawlings BJ, Reese PB, Ramer SE, Vederas JC. J. Am. Chem. Soc. 1989; 111: 3382
- 46a Martín VS, Nuñez MT, Ramírez MA, Soler MA. Tetrahedron Lett. 1990; 31: 763
- 46b Palazón JM, Soler MA, Ramírez MA, Martín VS. Tetrahedron Lett. 1993; 34: 5467 ; see also ref. 28d
- 47 Martín VS, Nuñez T, Tonn CE. Tetrahedron Lett. 1988; 29: 2701
- 48 Ando K. Tetrahedron Lett. 1995; 36: 4105
- 49 Ramírez MA, Padrón JM, Palazón JM, Martín VS. J. Org. Chem. 1997; 62: 4584
- 50 Hoffmann RW. Angew. Chem. Int. Ed. Engl. 1992; 31: 1124
- 51 Betancort JM, Martín VS, Padrón JM, Palazón JM, Ramírez MA, Soler MA. J. Org. Chem. 1997; 62: 4570
- 52 Soler MA, Palazón JM, Martín VS. Tetrahedron Lett. 1993; 34: 5471
- 53 Nicholas KM. Acc. Chem. Res. 1987; 20: 207
- 54 For early synthetic applications of the Nicholas reaction from our group, see: Díaz DD, Betancort JM, Martín VS. Synlett 2007; 343
- 55 Crisóstomo FR. P, Martín T, Martín VS. Org. Lett. 2004; 6: 565
- 56 Takahashi S, Kubota A, Nakata T. Tetrahedron 2003; 59: 1627
- 57 Crisóstomo FR. P, Carrillo R, León LG, Martín T, Padrón JM, Martín VS. J. Org. Chem. 2006; 71: 2339
- 58a Xiong Z, Corey EJ. J. Am. Chem. Soc. 2000; 122: 9328
- 58b Mi Y, Schreiber JV, Corey EJ. J. Am. Chem. Soc. 2002; 124: 11290
- 59a McDonald FE, Wang X, Do B, Hardcastle KI. Org. Lett. 2000; 2: 2917
- 59b McDonald FE, Bravo F, Wang X, Wei X, Toganoh M, Rodríguez JR, Do B, Neiwert WA, Hardcastle KI. J. Org. Chem. 2002; 67: 2515
- 59c Xiong Z, Busch R, Corey EJ. Org. Lett. 2010; 12: 1512
- 60 Tanuwidjaja J, Ng S.-S, Jamison TF. J. Am. Chem. Soc. 2009; 131: 12084
- 61a Lindel T, Franck B. Tetrahedron Lett. 1995; 36: 9465
- 61b Enders D, Schüßeler T. Synthesis 2002; 2280
- 62 Bravo F, McDonald FE, Neiwert WA, Do B, Hardcastle KI. Org. Lett. 2003; 5: 2123
- 63 Johnson WS, Werthemann L, Bartlett WR, Brocksom TJ, Li T.-T, Faulkner DJ, Petersen MR. J. Am. Chem. Soc. 1970; 92: 741
- 64 Crisóstomo FR. P, Carrillo R, Martín T, García-Tellado F, Martín VS. J. Org. Chem. 2005; 70: 10099
- 65 Wong OA, Shi Y. Chem. Rev. 2008; 108: 3958
- 66 Rodríguez-López J, Crisóstomo FP, Ortega N, López-Rodríguez M, Martín VS, Martín T. Angew. Chem. Int. Ed. 2013; 52: 3659
- 67a Palazón JM, Martín VS. Tetrahedron Lett. 1995; 36: 3549
- 67b Díaz DD, Martín T, Martín VS. Org. Lett. 2001; 3: 3289
- 67c Díaz DD, Betancort JM, Crisóstomo FR. P, Martín T. Tetrahedron 2002; 58: 1913
- 68 Betancort JM, Martín T, Palazón JM, Martín VS. J. Org. Chem. 2003; 68: 3216
- 69a Ortega N, Martín T, Martín VS. Org. Lett. 2006; 8: 871
- 69b Ortega N, Martín T, Martín VS. Eur. J. Org. Chem. 2009; 554
- 70 For representative examples of enyne metathesis, see: Diver ST, Giessert AJ. Chem. Rev. 2004; 104: 1317
- 71a Green JR. Synlett 2001; 353
- 71b Burlison JA, Gray JM, Young DG. J. Tetrahedron Lett. 2001; 42: 5363
- 71c Yang Z.-Q, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. J. Am. Chem. Soc. 2004; 126: 7881
- 72 Crisóstomo FR. P, Carrillo R, Martín T, Martín VS. Tetrahedron Lett. 2005; 46: 2829
- 73 Ortega N, Martín VS, Martín T. J. Org. Chem. 2010; 75: 6660
- 74 Irie T, Suzuki M, Masamune T. Tetrahedron Lett. 1965; 6: 1091
- 75 Bratz M, Bullock WH, Overman LE, Takemoto T. J. Am. Chem. Soc. 1995; 117: 5958
- 76a Sharpless KB, Young MW. J. Org. Chem. 1974; 40: 947
- 76b Burke SD, Voight EA. Org. Lett. 2001; 237
- 77a Marumoto S, Jaber JJ, Vitale JP, Rychnovsky SD. Org. Lett. 2002; 4: 3919
- 77b Pastor IM, Yus M. Curr. Org. Chem. 2007; 11: 925
- 77c Pastor IM, Yus M. Curr. Org. Chem. 2012; 16: 1277
- 78 Miranda PO, Díaz DD, Padrón JI, Bermejo J, Martín VS. Org. Lett. 2003; 5: 1979
- 79 Miranda PO, Carballo RM, Ramírez MA, Martín VS, Padrón JI. ARKIVOC 2007; (iv): 331
- 80 Miranda PO, Padrón JM, Padrón JI, Villar J, Martín VS. ChemMedChem 2006; 1: 323
- 81 Miranda PO, Ramírez MA, Padrón JI, Martín VS. Tetrahedron Lett. 2006; 47: 283
- 82 Miranda PO, Ramírez MA, Padrón JI, Martín VS. Org. Lett. 2006; 8: 1633
- 83a Jasti R, Anderson CD, Rychnovsky SD. J. Am. Chem. Soc. 2005; 127: 9939
- 83b Jasti R, Rychnovsky SD. J. Am. Chem. Soc. 2006; 128: 13640
- 84 Miranda PO, Ramírez MA, Martín VS, Padrón JI. Chem. Eur. J. 2008; 14: 6260
- 85a Onishi Y, Ito T, Yasuda M, Baba A. Eur. J. Org. Chem. 2002; 1578
- 85b Onishi Y, Ito T, Yasuda M, Baba A. Tetrahedron 2002; 58: 8227
- 85c Chan K.-P, Loh T.-P. Org. Lett. 2005; 7: 4491
- 85d Chan K.-P, Seow A.-H, Loh T.-P. Tetrahedron Lett. 2007; 48: 37
- 85e Chan K.-P, Ling YH, Loh T.-P. Chem. Commun. 2007; 939
- 85f Liu F, Loh T.-P. Org. Lett. 2007; 9: 2063
- 86 Miranda PO, Carballo RM, Martín VS, Padrón JI. Org. Lett. 2009; 11: 357
- 87a Coppi L, Ricci A, Taddei M. J. Org. Chem. 1988; 53: 913
- 87b Lolkema LD. M, Hiemstra H, Mooiweer HH, Speckamp WN. Tetrahedron Lett. 1988; 29: 6365
- 87c Furman B, Dziedzic M, Justyniak I. Tetrahedron 2008; 64: 3103
- 88a Overman LE, Castañeda A, Blumenkopf TA. J. Am. Chem. Soc. 1986; 108: 1303
- 88b Castañeda A, Kucera DJ, Overman LE. J. Org. Chem. 1989; 54: 5695
- 88c Berger D, Overman LE, Renhowe PA. J. Am. Chem. Soc. 1997; 119: 2446
- 89 Purino MA, Ramírez MA, Daranas AH, Martín VS, Padrón JI. Org. Lett. 2012; 14: 5904
- 90 The relative stereochemistry of the tetrahydrofurans was determined by means of NMR spectroscopy, see: Napolitano JG, Gavín JA, García C, Norte M, Fernández JJ, Daranas AH. Chem. Eur. J. 2011; 17: 6338
For preliminary results, see: