

Subscribe to RSS
DOI: 10.1055/s-0033-1340044
An Unprecedented Ring Transformation of a 4-(Aminomethyl)oxazoline Derivative to a 4-(Hydroxymethyl)imidazoline
Authors
Publication History
Received: 27 August 2013
Accepted after revision: 30 September 2013
Publication Date:
04 November 2013 (online)

Abstract
An optically active 4-(azidomethyl)-2-(2-pyridyl)oxazoline was prepared starting from l-serine and picolinic acid. Reduction of the azide moiety gave the corresponding 4-(aminomethyl)-2-(2-pyridyl)oxazoline, which is not a stable compound but readily undergoes ring-transformation rearrangement to furnish a 4-(hydroxymethyl)-2-(2-pyridyl)imidazoline. After Boc protection of the amidine function, the material could be further converted into the 4-(aminomethyl)-2-(2-pyridyl)imidazoline via the respective azidomethyl compound.
Since the first reports by Brunner and co-workers,[1] optically active 2-pyridyloxazolines have become a privileged class of chiral ligands in asymmetric catalysis.[2] [3] In continuation of our earlier work on C 1-symmetric, tridentate pyridyloxazolines,[4] we were planning to prepare compound 1 with an aminomethyl group, the latter being perfectly suited for further N-functionalization, for example by amidation reactions. In the event, we were unable to isolate compound 1, nor its derivatives, as it always underwent ring transformation to furnish the hydroxymethyl-functionalized pyridylimidazoline 2 (Scheme [1]). Such a ring transformation has been reported once before for the formation of a cyclic urea from a urethane.[5] It has been at least suspected for [(N-arylamino)methyl]oxazolines,[6] but not observed by others, although some researchers claim to have isolated an (aminomethyl)oxazoline as an intermediate product.[7] In one other case, an in situ formed (aminomethyl)oxazoline was protected with CbzCl in the reaction mixture.[8] Since optically active imidazolines are an increasingly important, novel class of chiral ligands,[9] investigations on further conversion of the product 2 seemed promising to us. Therefore, we now report on the preparation of compound 1 and its transformation to imidazoline 2, as well as some further chemistry starting with the latter compound.
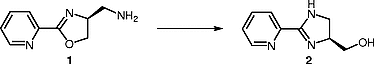
The starting point of our investigation was the hydroxymethyl compound 7, which was obtained by the five-step sequence outlined in Scheme [2]. Picolinic acid (3) was first coupled with methyl l-serinate[10] to give amide 4 (71%).[11] The primary alcohol was then TBS protected (92% yield of compound 5)[12] and the ester moiety reduced to again give a primary alcohol 8 (79%), which was activated with tosyl chloride and cyclized according to a literature protocol.[13] Intermediate product 6 (77%) was finally deprotected with tetrabutylammonium fluoride (93% yield of product 7).

Organoazides are excellent precursors for primary amines; therefore, we activated the primary alcohol function of compound 7 by sulfonyl ester formation (95% of product 9) and converted it with sodium azide in ethanol following common protocols (Scheme [3]).[14] Product 10, after chromatography, was obtained in almost quantitative yield. Azide 10 was subjected to catalytic hydrogenation to again give a single compound (95% yield) with correct mass (ESI) and 1H and 13C NMR spectra that seemed to be in agreement with structure 1; however, all attempts at further derivatization of what we assumed to be an amino function, in particular amide formation, failed. We realized that the ring-transformation reaction of intermediate product 1 with formation of imidazoline 2 would be an explanation for our failure to produce amides of purported amine 1. Imidazoline 2 would give similar 1H and 13C NMR spectra, leading us astray. This rearrangement appears obvious, since the amino group as a good nucleophile is at the right distance to form a bicyclo[2.2.1] intermediate structure when adding to the electrophilic imidoester group.

Definite proof of the structure of compound 2 came from its subsequent chemistry, which was initially difficult as the very basic amidine function caused several problems with chemoselectivity. These problems could, however, be solved by reducing the electron density of 2 by installing a carbamate function (product 11, 67% yield of crude material; Scheme [4]). Interestingly, we obtained a single compound, although one would expect a regioselectivity problem due to the presence of two nucleophilic nitrogen atoms at the amidine function. The depicted regiochemistry was proven by 2D-NMR spectroscopy, after the assignment of all resonances. Firstly, C-4 and 4-H were identified by DEPT135 and HMQC experiments [δ(13C) = 66.8, δ(1H) = 4.28 ppm]. Furthermore, the pyridine signals were identified by H,H-COSY and HMQC experiments, as follows [δ(1H), δ(13C), in ppm]: 7.5 (3′-H), 123.4 (C-3′); 7.7 (4′-H), 136.3 (C-4′); 7.29 (5′-H), 124.3 (C-5′); 8.58 (6′-H), 148.8 (C-6′). The quaternary pyridine C-2′ was then identified at 151.4 ppm (HMBC cross peaks with 6′-H and 4′-H). Of the two remaining sp2-carbon resonances, the amidine C-2 was assigned at δ = 159.4 ppm by the HMBC cross peak with 3′-H. Therefore, the carbamate C=O must be the remaining sp2 signal at δ = 150.4 ppm. The two methylene protons 5-H were then identified at δ = 3.84 and 3.99 ppm by the HMBC cross peak with the amidine C-2; the respective C-5 was identified at δ = 49.3 ppm (HMQC experiment). The remaining 4-CH2OH was identified at δ(1H) = 3.64 and 3.74 ppm, and δ(13C) = 64.3 ppm (HMQC experiment). With this assignment of all proton and carbon resonances in hand, the structure of compound 11 was finally proven by 15N,1H-HMBC spectroscopy; three 15N signals were observed, and were assigned according to the literature data for a similar 1-acetyl-2-aryl-4,5-dihydroimidazole:[15] δ(15N) = –74.1 (N-1′), –122.8 (N-3), –244.5 (N-1) ppm, with MeNO2 (δ = 0) as external standard. Naturally, within the pyridine ring, N-1′ showed cross peaks to 3′-H, 5′-H and 6′-H. The carbamate N-1 showed three strong cross peaks, to 4-H and both 5-H protons, whereas the imine N-3 showed two strong cross peaks, to both 4-CH 2OH protons, and one weak cross peak, to one 5-H proton.

The alcohol function of 11 could then be activated by tosylate formation (40% yield of product 12) and subsequently be displaced with azide to furnish compound 13 (73% yield). Finally, the aminomethyl compound 14 was obtained by hydrogenation of azide 13 (67% yield), and actually represents the N-Boc-imidazoline congener of our initial target compound 1. This material 14 will now be the cornerstone in future efforts for the synthesis of new libraries of chiral polydentate ligands.
In summary, during our studies towards new, optically active tridentate ligands, we considered aminomethyl-substituted pyridyloxazoline 1 as a core structure which would be ready for further derivatization at the primary amino function. However, attempts to prepare target compound 1 by reduction of the respective azidomethyl precursor 10 directly led to the rearranged product 2 with a hydroxymethyl-substituted imidazoline ring. This very basic amidine moiety hindered subsequent chemistry and was therefore protected with a tert-butoxycarbonyl group. It was then possible to access the (aminomethyl)(pyridyl)imidazoline 14 which is now ready for further diversifying derivatization at the primary amino function.
Preparative column chromatography was carried out using Merck silica gel (35–70 μm, type 60 A) with hexane, EtOAc and MeOH as eluents. The column dimensions are given as follows: diameter × height. TLC was performed on Merck aluminum plates coated with silica gel F254. 1H, 13C and 15N NMR spectra were recorded on a Bruker Avance DRX 500 instrument. Multiplicities of carbon signals were determined with DEPT experiments. MS and HRMS spectra were obtained with a Waters Q-Tof Premier (ESI) spectrometer. IR spectra were recorded on a Bruker Tensor 27 spectrometer equipped with a Golden Gate diamond ATR unit. Elemental analyses were determined with a Euro EA-CHNS instrument from HEKAtech. Optical rotations were measured with a Polartronic M polarimeter from Schmidt and Haensch. Methyl l-serinate hydrochloride was prepared according to a literature procedure.[10] All other starting materials were commercially available.
Methyl (S)-N-(2-Pyridylcarbonyl)serinate (4)
N-Methylmorpholine (10.3 mL, 9.46 g, 93.5 mmol) and ethyl chloroformate (4.20 mL, 4.85 g, 44.7 mmol) were added to an ice-cooled suspension of picolinic acid (3) (5.00 g, 40.6 mmol) in anhydrous THF (50 mL) and the mixture was stirred for 0.5 h at this temperature. Methyl l-serinate hydrochloride (7.00 g, 44.7 mmol) was added and, after further stirring for 2 h at r.t., the solvent was removed under reduced pressure. The residue was partitioned between EtOAc (50 mL) and H2O (50 mL), and the aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 7 cm × 11 cm; EtOAc–hexane, 2:1, Rf = 0.25) to yield compound 4 (6.46 g, 28.8 mmol, 71%) as a colorless liquid. All spectroscopic data were in accordance with the literature.[11]
[α]D 20 +38.9 (c 1.03, CH2Cl2) [Lit.[11] [α] D 21 +39.4 (c 2.5, CHCl3)].
1H NMR (500 MHz, CDCl3): δ = 2.87 (t, J = 5.7 Hz, 1 H, OH), 3.82 (s, 3 H), 4.03–4.14 (m, 2 H), 4.87 (dt, J = 7.7, 3.9 Hz, 1 H), 7.44 (ddd, J = 7.6, 4.8, 1.0 Hz, 1 H), 7.84 (td, J = 7.7, 1.7 Hz, 1 H), 8.16 (dt, J = 7.9, 1.0 Hz, 1 H), 8.58 (ddd, J = 4.8, 1.5, 0.9 Hz, 1 H), 8.80–8.85 (m, 1 H, NH).
13C{1H} NMR (125 MHz, CDCl3): δ = 52.8 (CH), 54.9 (CH3), 63.5 (CH2), 122.4 (CH), 126.5 (CH), 137.3 (CH), 148.3 (CH), 149.1 (C), 164.7 (C), 170.7 (C).
Methyl (S)-O-(tert-Butyldimethylsilyl)-N-(2-pyridylcarbonyl)serinate (5)
TBSCl (4.81 g, 31.9 mmol) and imidazole (5.00 g, 73.5 mmol) were added to a solution of compound 4 (5.49 g, 24.5 mmol) in CH2Cl2 (70 mL), and the mixture was stirred for 1 h at r.t. and then filtered. The solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel, 7 cm × 11 cm; EtOAc–hexane, 1:2, Rf = 0.40) to yield compound 5 (7.66 g, 22.6 mmol, 92%) as a colorless liquid.
[α]D 20 +35.3 (c 1.13, CH2Cl2).
IR (ATR): 3396 (w), 2953 (w), 2930 (w), 2885 (w), 2857 (w), 1749 (m), 1680 (m), 1592 (w), 1571 (w), 1512 (s), 1465 (m), 1435 (m), 1380 (w), 1352 (m), 1292 (w), 1253 (m), 1207 (m), 1168 (m), 1107 (s), 1043 (m), 998 (m), 965 (w), 939 (w) cm–1.
1H NMR (500 MHz, CDCl3): δ = 0.009 (s, 3 H), 0.011 (s, 3 H), 0.89 (s, 9 H), 3.75 (s, 3 H), 3.95 (dd, J = 10.1, 3.5 Hz, 1 H), 4.19 (dd, J = 10.1, 3.0 Hz, 1 H), 4.86 (dt, J = 8.6, 3.2 Hz, 1 H), 7.43 (ddd, J = 7.6, 4.7, 1.2 Hz, 1 H), 7.84 (td, J = 7.7, 1.7 Hz, 1 H), 8.18 (dt, J = 7.8, 1.1 Hz, 1 H), 8.60 (ddd, J = 4.8, 1.6, 0.8 Hz, 1 H), 8.74–8.78 (m, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = –5.6 (CH3), –5.5 (CH3), 18.3 (C), 25.8 (3 CH3), 52.5 (CH), 54.6 (CH3), 63.8 (CH2), 122.4 (CH), 126.4 (CH), 137.3 (CH), 148.5 (CH), 149.7 (C), 164.3 (C), 170.9 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C16H26N2NaO4Si: 361.1560; found: 361.1552.
Anal. Calcd for C16H26N2O4Si (338.48): C, 56.78; H, 7.74; N, 8.28. Found: C, 56.77; H, 7.82; N, 8.40.
N-[(R)-2-(tert-Butyldimethylsilyloxy)-1-(hydroxymethyl)ethyl]picolinamide (8)
Compound 5 (9.48 g, 28.0 mmol) was added dropwise to an ice-cooled solution of LiBH4 (18.3 mL of a 2 M solution in THF, 36.6 mmol; diluted with 18.3 mL anhydrous THF) and the mixture was stirred for 3 h at this temperature. Subsequently, H2O (10 mL) and 10% HCl (ca. 20 mL) were added with caution. After gas evolution had stopped, sat. aq NaHCO3 solution (ca. 40 mL) was added and the mixture was extracted with EtOAc (4 × 40 mL). The combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 7 cm × 12 cm; EtOAc–hexane, 1:1, Rf = 0.20) to yield compound 8 (6.88 g, 22.2 mmol, 79%) as a colorless liquid.
[α]D 20 +3.33 (c 1.00, CH2Cl2).
IR (ATR): 3384 (m), 2955 (m), 2931 (m), 2885 (w), 2859 (m), 1666 (s), 1593 (w), 1572 (w), 1521 (s), 1466 (m), 1436 (m), 1391 (w), 1363 (w), 1293 (w), 1255 (m), 1091 (s), 1046 (m), 1022 (m), 1000 (m), 940 (w), 835 (s), 779 (s) cm–1.
1H NMR (500 MHz, CDCl3): δ = 0.08 (s, 3 H), 0.09 (s, 3 H), 0.92 (s, 9 H), 3.01 (dd, J = 7.8, 4.0 Hz, 1 H), 3.80–3.85 (m, 1 H), 3.86–3.98 (m, 3 H), 4.13–4.20 (m, 1 H), 7.42 (ddd, J = 7.7, 4.7, 1.2 Hz, 1 H), 7.85 (td, J = 7.7, 1.6 Hz, 1 H), 8.19 (dt, J = 7.8, 1.0 Hz, 1 H), 8.56 (ddd, J = 4.8, 1.6, 0.8 Hz, 1 H), 8.63–8.67 (m, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = –5.6 (CH3), –5.4 (CH3), 18.3 (C), 26.0 (3 CH3), 52.5 (CH), 63.8 (CH2), 64.2 (CH2), 122.3 (CH), 126.3 (CH), 137.4 (CH), 148.4 (CH), 150.0 (C), 164.9 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C15H26N2NaO3Si: 333.1610; found: 333.1612.
Anal. Calcd for C15H26N2O3Si (310.47): C, 58.03; H, 8.44; N, 9.02. Found: C, 58.01; H, 8.44; N, 9.02.
(R)-4-(tert-Butyldimethylsilyloxymethyl)-2-(2-pyridyl)-4,5-dihydrooxazole (6)
TsCl (1.47 g, 7.73 mmol), DMAP (78 mg, 0.64 mmol) and Et3N (4.48 mL, 3.26 g, 32.2 mmol) were added to a solution of compound 8 (2.00 g, 6.44 mmol) in DCE (40 mL) and the resulting mixture was heated to reflux for 16 h. The organic layer was washed with H2O (3 × 30 mL), dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 5 cm × 12 cm; EtOAc–hexane, 1:1, Rf = 0.20) to yield compound 6 (1.45 g, 4.96 mmol, 77%) as a colorless liquid.
[α]D 20 –37.5 (c 0.99, CH2Cl2).
IR (ATR): 2955 (m), 2930 (m), 2899 (w), 2858 (m), 1643 (m), 1585 (w), 1572 (w), 1519 (w), 1473 (m), 1442 (m), 1363 (m), 1253 (m), 1109 (m), 1045 (m), 1009 (m), 967 (m), 940 (w), 897 (w), 835 (s), 803 (m), 777 (s), 746 (m) cm–1.
1H NMR (500 MHz, CDCl3): δ = 0.04 (s, 3 H), 0.07 (s, 3 H), 0.86 (s, 9 H), 3.65–3.70 (m, 1 H), 3.94 (dd, J = 10.2, 3.4 Hz, 1 H), 4.42–4.50 (m, 2 H), 4.51–4.57 (m, 1 H), 7.38 (ddd, J = 7.6, 4.8, 1.1 Hz, 1 H), 7.77 (td, J = 7.8, 1.6 Hz, 1 H), 8.03 (dt, J = 7.9, 1.0 Hz, 1 H), 8.70 (ddd, J = 4.8, 1.7, 0.9 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = –5.2 (2 CH3), 18.4 (C), 26.0 (3 CH3), 65.2 (CH2), 68.7 (CH), 71.2 (CH2), 124.0 (CH), 125.7 (CH), 136.7 (CH), 147.0 (C), 149.9 (CH), 164.1 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C15H24N2NaO2Si: 315.1505; found: 315.1500.
Anal. Calcd for C15H24N2O2Si (292.45): C, 61.60; H, 8.27; N, 9.58. Found: C, 61.59; H, 8.29; N, 9.52.
(S)-4-(Hydroxymethyl)-2-(2-pyridyl)-4,5-dihydrooxazole (7)
TBAF·3H2O (1.94 g, 6.16 mmol) was added to a solution of compound 6 (1.50 g, 5.13 mmol) in THF (50 mL) and the resulting mixture was stirred for 1 h at r.t. The solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel, 5 cm × 10 cm; EtOAc–MeOH, 5:1, Rf = 0.24) to yield compound 7 (0.850 g, 4.77 mmol, 93%) as a colorless oil.
[α]D 20 –61.6 (c 0.69, MeOH).
IR (ATR): 3288 (m), 2982 (w), 2927 (w), 2874 (w), 1664 (m), 1582 (m), 1483 (m), 1432 (m), 1398 (w), 1381 (m), 1360 (s), 1343 (m), 1301 (m), 1285 (m), 1270 (m), 1247 (m), 1222 (m), 1199 (w), 1155 (w), 1125 (s), 1098 (m), 1086 (m), 1070 (s), 1042 (m), 995 (m), 983 (m), 952 (s), 933 (m), 908 (w), 890 (m) cm–1.
1H NMR (500 MHz, CDCl3): δ = 3.18 (s, 1 H), 3.70 (dd, J = 11.6, 4.0 Hz, 1 H), 4.01 (dd, J = 11.6, 3.4 Hz, 1 H), 4.42–4.46 (m, 1 H), 4.47–4.53 (m, 1 H), 4.57 (dd, J = 9.5, 7.2 Hz, 1 H), 7.35 (ddd, J = 7.6, 4.8, 0.9 Hz, 1 H), 7.73 (td, J = 7.7, 1.7 Hz, 1 H), 7.93 (dt, J = 7.9, 1.2 Hz, 1 H), 8.64 (ddd, J = 4.8, 1.6, 0.9 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = 64.0 (CH2), 68.6 (CH), 70.0 (CH2), 124.0 (CH), 125.9 (CH), 136.8 (CH), 146.2 (C), 149.9 (CH), 164.5 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C9H10N2NaO2: 201.0640; found: 201.0635.
(R)-4-[(4-Methylphenylsulfonyloxy)methyl]-2-(2-pyridyl)-4,5-dihydrooxazole (9)
TsCl (1.07 g, 5.60 mmol) was added to a solution of compound 7 (0.665 g, 3.73 mmol) in 15% aq KOH solution (25 mL) and CH2Cl2 (25 mL), and the resulting mixture was heated to reflux for 3 h. The layers were separated and the aqueous layer was extracted with CH2Cl2 (50 mL). The combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 5 cm × 8 cm; EtOAc–MeOH, 5:1, Rf = 0.40) to yield compound 9 (1.18 g, 3.56 mmol, 95%) as a colorless solid; mp 113 °C.
[α]D 20 –82.9 (c 1.15, CH2Cl2).
IR (ATR): 3055 (w), 2968 (w), 2902 (w), 2324 (w), 1732 (w), 1645 (m), 1599 (m), 1568 (m), 1494 (w), 1471 (m), 1442 (m), 1348 (m), 1306 (m), 1292 (m), 1280 (m), 1246 (m), 1213 (m), 1175 (s), 1121 (m), 1098 (m), 1080 (m), 1039 (m), 1012 (m), 994 (m), 966 (m), 948 (s), 932 (m), 898 (m), 866 (m) cm–1.
1H NMR (500 MHz, CDCl3): δ = 2.41 (s, 3 H), 4.05 (dd, J = 10.0, 6.6 Hz, 1 H), 4.29 (dd, J = 10.0, 3.7 Hz, 1 H), 4.36 (dd, J = 8.1, 6.7 Hz, 1 H), 4.51–4.62 (m, 2 H), 7.30 (d, J = 8.2 Hz, 2 H), 7.39 (ddd, J = 7.7, 4.7, 1.1 Hz, 1 H), 7.72–7.79 (m, 3 H), 7.91–7.94 (m, 1 H), 8.68 (ddd, J = 4.9, 1.8, 0.9 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = 21.7 (CH3), 65.5 (CH), 70.5 (CH2), 70.6 (CH2), 124.2 (CH), 126.1 (CH), 128.1 (2 CH), 130.0 (2 CH), 132.6 (C), 136.8 (CH), 145.2 (C), 146.1 (C), 150.0 (CH), 165.0 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C16H16N2NaO4S: 355.0728; found: 355.0728.
Anal. Calcd for C16H16N2O4S (332.37): C, 57.82; H, 4.85; N, 8.43; S, 9.65. Found: C, 57.91; H, 4.86; N, 8.47; S, 9.65.
(S)-4-(Azidomethyl)-2-(2-pyridyl)-4,5-dihydrooxazole (10)
NaN3 (3.39 g, 52.1 mmol) was added to a solution of compound 9 (3.46 g, 10.4 mmol) in EtOH (150 mL) and the resulting mixture was heated to reflux for 16 h. After the solvent was removed, the residue was partitioned between CH2Cl2 (100 mL) and H2O (100 mL), and the aqueous layer was extracted with CH2Cl2 (50 mL). The combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 5 cm × 10 cm; EtOAc–hexane, 9:1, Rf = 0.15) to yield compound 10 (2.12 g, 10.4 mmol, 99%) as a light yellow oil.
[α]D 20 –112.5 (c 1.01, CH2Cl2).
IR (ATR): 3057 (w), 2969 (w), 2902 (w), 2096 (vs), 1639 (m), 1581 (m), 1569 (m), 1516 (w), 1469 (m), 1440 (m), 1366 (m), 1258 (m), 1151 (w), 1102 (s), 1066 (m), 1043 (m), 994 (m), 965 (m), 931 (m), 905 (m), 856 (w), 800 (m) cm–1.
1H NMR (500 MHz, CDCl3): δ = 3.53–3.62 (m, 2 H), 4.33–4.41 (m, 1 H), 4.54–4.62 (m, 2 H), 7.42 (ddd, J = 7.6, 4.8, 1.0 Hz, 1 H), 7.80 (td, J = 7.8, 1.7 Hz, 1 H), 8.05 (dt, J = 7.9, 1.0 Hz, 1 H), 8.72 (ddd, J = 4.8, 1.6, 1.0 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = 54.2 (CH2), 66.5 (CH), 70.7 (CH2), 124.2 (CH), 125.9 (CH), 136.7 (CH), 146.2 (C), 149.9 (CH), 164.5 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C9H9N5NaO: 226.0705; found: 226.0699.
Anal. Calcd for C9H9N5O (203.21): C, 53.20; H, 4.46; N, 34.37. Found: C, 53.16; H, 4.47; N, 34.29.
(S)-4-(Hydroxymethyl)-2-(2-pyridyl)-4,5-dihydro-1H-imidazole (2)
A suspension of compound 10 (2.21 g, 10.9 mmol) and Pd/C (221 mg, 10% w/w Pd) in MeOH (50 mL) was degassed (three cycles of freeze, pump and thaw) and then stirred under an atmosphere of H2 (1 atm) for 16 h at 23 °C. The mixture was filtered, then the solvent was removed under reduced pressure to yield compound 2 (1.82 g, 10.3 mmol, 95%) as a yellow oil.
[α]D 20 +55.3 (c 1.11, MeOH).
IR (ATR): 3279 (br), 2927 (m), 2862 (m), 2453 (w), 1659 (w), 1601 (m), 1566 (m), 1487 (m), 1456 (m), 1421 (m), 1331 (m), 1271 (m), 1189 (m), 1096 (m), 1045 (m), 979 (m), 801 (m) cm–1.
1H NMR (500 MHz, CD3OD): δ = 3.61 (dd, J = 11.0, 5.6 Hz, 1 H), 3.65 (dd, J = 11.1, 5.5 Hz, 1 H), 3.70 (dd, J = 12.3, 7.5 Hz, 1 H), 3.92 (dd, J = 12.2, 10.9 Hz, 1 H), 4.19 (ddt, J = 10.7, 7.5, 5.6 Hz, 1 H), 7.50 (ddd, J = 7.5, 4.9, 1.0 Hz, 1 H), 7.90 (td, J = 7.8, 1.7 Hz, 1 H), 8.02 (dt, J = 7.9, 1.0 Hz, 1 H), 8.63 (ddd, J = 4.9, 1.5, 1.1 Hz, 1 H).
13C{1H} NMR (125 MHz, CD3OD): δ = 53.5 (CH2), 63.6 (CH), 65.5 (CH2), 123.5 (CH), 126.9 (CH), 138.2 (CH), 148.8 (C), 150.2 (CH), 165.5 (C).
HRMS (ESI, positive mode): m/z [M + H+] calcd for C9H12N3O: 178.0980; found: 178.0975.
Anal. Calcd for C9H11N3O (177.21): C, 61.00; H, 6.26; N, 23.71. Found: C, 60.89; H, 6.57; N, 23.73.
(S)-1-(tert-Butoxycarbonyl)-4-(hydroxymethyl)-2-(2-pyridyl)-4,5-dihydro-1H-imidazole (11)
K2CO3 (2.70 g, 19.5 mmol) and Boc2O (5.33 g, 24.4 mmol) were added to a solution of compound 2 (1.73 g, 9.76 mmol) in H2O–THF (30 mL, 1:3) and the resulting mixture was heated to reflux for 24 h. All volatile materials were then removed under reduced pressure, the residue was partitioned between CH2Cl2 (100 mL) and H2O (100 mL), the aqueous layer was extracted with CH2Cl2 (50 mL), and the combined organic layers were dried (MgSO4) and filtered. The solvent was removed under reduced pressure to yield compound 11 as a mixture with Boc2O (2.30 g) as crude material (Boc2O/11, 1:3 by 1H NMR spectroscopy; i.e., 1.82 g, 6.57 mmol, 67%), which was used for the next step without further purification. An analytically pure sample was obtained by column chromatography (silica gel, 3 cm × 10 cm; EtOAc–MeOH, 5:1, Rf = 0.22) as a light yellow oil.
[α]D 20 +92.5 (c 0.46, CH2Cl2).
IR (ATR): 3312 (br), 2979 (w), 2932 (w), 2871 (w), 1708 (s), 1631 (m), 1588 (w), 1476 (w), 1365 (vs), 1286 (m), 1138 (vs), 1049 (w), 1025 (w), 917 (w), 729 (s) cm–1.
1H NMR (500 MHz, CDCl3): δ = 1.21 (s, 9 H, 3 CH3), 3.64 (dd, J = 11.4, 5.0 Hz, 1 H, 4-CHH), 3.74 (dd, J = 11.4, 4.5 Hz, 1 H, 4-CHH), 3.84 (dd, J = 10.5, 8.0 Hz, 1 H, 5-H), 3.99 (t, J = 10.4 Hz, 1 H, 5-H), 4.28 (ddt, J = 10.3, 8.1, 4.7 Hz, 1 H, 4-H), 7.29 (ddd, J = 7.7, 4.9, 1.1 Hz, 1 H, 5′-H), 7.47–7.50 (m, 1 H, 3′-H), 7.70 (td, J = 7.7, 1.7 Hz, 1 H, 4′-H), 8.58 (ddd, J = 4.9, 1.7, 1.1 Hz, 1 H, 6′-H).
13C{1H} NMR (125 MHz, CDCl3): δ = 27.8 (3 CH3, C(CH3 ) 3), 49.3 (CH2, C-5), 64.3 (CH2, 4-CH2), 66.8 (CH, C-4), 81.9 (C, C(CH3 ) 3), 123.4 (CH, C-3′), 124.3 (CH, C-5′), 136.3 (CH, C-4′), 148.8 (CH, C-6′), 150.4 (C, C=O), 151.4 (C, C-2′), 159.4 (C, C-2).
15N NMR (HMBC, 50.7 MHz, CDCl3): δ = –74.1 (N-1′), –122.8 (N-3), –244.5 (N-1).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C14H19N3NaO3: 300.1324; found: 300.1319.
(S)-1-(tert-Butoxycarbonyl)-4-[(4-methylphenylsulfonyloxy)methyl]-2-(2-pyridyl)-4,5-dihydro-1H-imidazole (12)
TsCl (7.23 g, 37.9 mmol) was added to a solution of crude compound 11 (2.10 g, Boc2O/11, 1:3 by 1H NMR spectroscopy; i.e., 1.66 g, 6.00 mmol) in 15% aq KOH solution (50 mL) and CH2Cl2 (50 mL), and the resulting mixture was heated to reflux for 3 h. The layers were separated and the aqueous layer was extracted with CH2Cl2 (50 mL). The combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure. The residue was purified by column chromatography (silica gel, 5 cm × 10 cm; EtOAc–MeOH, 5:1, Rf = 0.23) to yield compound 12 (1.03 g, 2.39 mmol, 40%; 27% over two steps) as a yellow oil.
[α]D 20 +65.8 (c 0.25, CH2Cl2).
IR (ATR): 2978 (w), 2931 (w), 2871 (w), 1708 (s), 1630 (w), 1588 (w), 1474 (w), 1365 (vs), 1288 (m), 1175 (vs), 1096 (m), 1049 (w), 982 (s), 950 (m), 815 (w), 790 (m), 665 (m), 554 (s) cm–1.
1H NMR (500 MHz, CDCl3): δ = 1.21 (s, 9 H), 2.43 (s, 3 H), 3.81 (dd, J = 11.1, 7.2 Hz, 1 H), 3.99–4.13 (m, 2 H), 4.27 (dd, J = 9.9, 4.1 Hz, 1 H), 4.36–4.48 (m, 1 H), 7.29–7.37 (m, 3 H), 7.46 (dt, J = 8.0, 0.9 Hz, 1 H), 7.72 (td, J = 7.8, 1.8 Hz, 1 H), 7.77–7.81 (m, 2 H), 8.61 (ddd, J = 4.9, 1.5, 1.1 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = 21.8 (CH3), 27.8 (3 CH3), 50.0 (CH2), 63.5 (CH), 70.9 (CH2), 82.4 (C), 123.4 (CH), 124.5 (CH), 128.2 (2 CH), 130.1 (2 CH), 132.6 (C), 136.4 (CH), 145.1 (C), 149.0 (CH), 150.2 (C), 151.1 (C), 160.5 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C21H25N3NaO5S: 454.1413; found: 454.1403.
(S)-4-(Azidomethyl)-1-(tert-butoxycarbonyl)-2-(2-pyridyl)-4,5-dihydro-1H-imidazole (13)
A solution of NaN3 (776 mg, 11.9 mmol) and compound 12 (1.03 g, 2.39 mmol) in EtOH (20 mL) was heated to reflux for 16 h. Then, the solvent was removed under reduced pressure and the residue was partitioned between CH2Cl2 (25 mL) and H2O (25 mL). The aqueous layer was extracted with CH2Cl2 (25 mL), the combined organic layers were dried (MgSO4) and filtered, and the solvent was removed under reduced pressure to yield compound 13 (530 mg, 1.75 mmol, 73%) as a yellow oil.
[α]D 20 +95.6 (c 0.79, CH2Cl2).
IR (ATR): 2979 (w), 2932 (w), 2102 (s), 1711 (s), 1633 (w), 1587 (w), 1520 (w), 1472 (w), 1368 (vs), 1277 (m), 1259 (m), 1168 (m), 1142 (s), 997 (w), 982 (s) cm–1.
1H NMR (500 MHz, CDCl3): δ = 1.20 (s, 9 H), 3.51 (dd, J = 12.4, 4.7 Hz, 1 H), 3.59 (dd, J = 12.4, 5.4 Hz, 1 H), 3.82 (dd, J = 10.9, 7.1 Hz, 1 H), 4.07 (t, J = 10.6 Hz, 1 H), 4.36–4.42 (m, 1 H), 7.33 (ddd, J = 7.7, 4.8, 1.1 Hz, 1 H), 7.53 (dt, J = 7.7, 1.0 Hz, 1 H), 7.74 (td, J = 7.7, 1.5 Hz, 1 H), 8.62 (ddd, J = 5.4, 1.5, 1.0 Hz, 1 H).
13C{1H} NMR (125 MHz, CDCl3): δ = 27.7 (3 CH3), 50.2 (CH2), 54.5 (CH2), 64.6 (CH), 82.1 (C), 123.2 (CH), 124.3 (CH), 136.3 (CH), 148.9 (CH), 150.3 (C), 151.4 (C), 159.9 (C).
HRMS (ESI, positive mode): m/z [M + Na+] calcd for C14H18N6NaO2: 325.1389; found: 325.1380.
(R)-4-(Aminomethyl)-1-(tert-butoxycarbonyl)-2-(2-pyridyl)-4,5-dihydro-1H-imidazole (14)
A suspension of compound 13 (208 mg, 0.69 mmol) and Pd/C (21 mg, 10% w/w Pd) in MeOH (20 mL) was degassed (three cycles of freeze, pump and thaw) and then stirred under an atmosphere of H2 (1 atm) for 16 h at 23 °C. The mixture was filtered, then the solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel, 3 cm × 5 cm; EtOAc–MeOH, 5:1, Rf = 0.08) to yield compound 14 (126 mg, 0.46 mmol, 67%) as a colorless oil.
[α]D 20 –27.8 (c 0.85, MeOH).
IR (ATR): 3333 (w), 3055 (w), 2976 (w), 2930 (w), 2867 (w), 1690 (s), 1607 (m), 1567 (m), 1494 (m), 1457 (m), 1421 (m), 1366 (s), 1273 (s), 1249 (s), 1163 (vs), 994 (w), 979 (w), 744 (w) cm–1.
1H NMR (500 MHz, CD3OD): δ = 1.33 (s, 9 H), 3.16–3.18 (m, 2 H), 3.57 (dd, J = 12.4, 7.2 Hz, 1 H), 3.84 (dd, J = 10.7, 7.1 Hz, 1 H), 4.14 (ddt, J = 10.9, 7.0, 5.7 Hz, 1 H), 7.42 (ddd, J = 7.6, 4.8, 1.1 Hz, 1 H), 7.53 (td, J = 7.7, 1.7 Hz, 1 H), 7.93 (dt, J = 7.9, 1.2 Hz, 1 H), 8.55 (ddd, J = 4.9, 1.6, 0.9 Hz, 1 H).
13C{1H} NMR (125 MHz, CD3OD): δ = 28.7 (3 CH3), 45.4 (CH2), 53.9 (CH2), 62.0 (CH), 80.2 (C), 123.7 (CH), 127.1 (CH), 138.3 (CH), 148.4 (C), 150.3 (CH), 158.7 (C), 165.6 (C).
HRMS (ESI, positive mode): m/z [M + H+] calcd for C14H21N4O2: 277.1665; found: 277.1659.
Acknowledgment
We are grateful to Andre Appeldorn for his assistance in the laboratory.
-
References
- 1a Brunner H, Obermann U, Wimmer P. J. Organomet. Chem. 1986; 316: C1
- 1b Brunner H, Obermann U. Chem. Ber. 1989; 122: 499
- 1c Brunner H, Obermann U, Wimmer P. Organometallics 1989; 8: 821
- 1d Brunner H, Brandl P. J. Organomet. Chem. 1990; 390: C81
- 1e Brunner H, Wutz K, Doyle MP. Monatsh. Chem. 1990; 121: 755
- 2a Desimoni G, Faita G, Quadrelli P. Chem. Rev. 2003; 103: 3119
- 2b Aspinall HC, Greeves N. J. Organomet. Chem. 2002; 647: 151
- 2c Johnson JS, Evans DA. Acc. Chem. Res. 2000; 33: 325
- 2d Nishiyama H, Ito J.-i. Chem. Commun. 2010; 46: 203
- 2e Ito J.-i, Nishiyama H. Synlett 2012; 23: 509
- 2f Ward BD, Gade L. Chem. Commun. 2012; 48: 10587
- 3a Ingalls EL, Sibbald PA, Kaminsky W, Michael FE. J. Am. Chem. Soc. 2013; 135: 8854
- 3b De Crisci AG, Chung K, Oliver AG, Solis-Ibarra D, Waymouth RM. Organometallics 2013; 32: 2257
- 3c Narayama A, Shibatomi K, Soga Y, Muto T, Iwasa S. Synlett 2013; 24: 375
- 3d Holder JC, Marziale AN, Gatti M, Mao B, Stoltz BM. Chem. Eur. J. 2013; 19: 74
- 3e Aguado-Ullate S, Urbano-Cuadrado M, Villalba I, Pires E, Garcia JI, Bo C, Carbo JJ. Chem. Eur. J. 2012; 18: 14026
- 3f Yang G, Shen C, Zhang W. Angew. Chem. Int. Ed. 2012; 51: 9141; Angew. Chem. 2012, 124, 9275
- 3g Malkov AV, Stewart-Liddon AJ. P, McGeoch GD, Ramirez-Lopez P, Kocovsky P. Org. Biomol. Chem. 2012; 10: 4864
- 4 Christoffers J, Mann A, Pickardt J. Tetrahedron 1999; 55: 5377
- 5 Maciagiewicz IM, Fang F, Roberts DA, Zhou S, Hines JV, Bergmeier SC. Synthesis 2012; 44: 551
- 6 Griffith JA, Rowlands GJ. Synthesis 2005; 3446
- 7 Chang Y.-w, Yang J.-j, Dang J.-n, Xue Y.-x. Synlett 2007; 2283
- 8 Cook GR, Shanker PS, Peterson SL. Org. Lett. 1999; 1: 615
Reference Ris Wihthout Link
- 9a Bhor S, Anilkumar G, Tse MK, Klawonn M, Döbler C, Bitterlich B, Grotevendt A, Beller M. Org. Lett. 2005; 7: 3393
- 9b Tursky M, Necas D, Drabina P, Sedlak M, Kotora M. Organometallics 2006; 25: 901
- 9c Ma K, You J. Chem. Eur. J. 2007; 13: 1863
- 9d Ohara M, Nakamura S, Shibata N. Adv. Synth. Catal. 2011; 353: 3285
- 9e Hyodo K, Nakamura S, Shibata N. Angew. Chem. Int. Ed. 2012; 51: 10337 ; Angew. Chem. 2012, 124, 10483
- 9f Barakat A, Islam MS, Al Majid AM. A, Al-Othman ZA. Tetrahedron 2013; 69: 5185
- 9g Weber M, Klein JE. M. N, Miehlich B, Frey W, Peters R. Organometallics 2013; 32: 5810
- 9h For a review, see: Liu H, Du D.-M. Adv. Synth. Catal. 2009; 351: 489
- 10 Huang Y, Dalton DR, Carroll PJ. J. Org. Chem. 1997; 62: 372
- 11 Merritt EA, Bagley MC. J. Heterocycl. Chem. 2007; 44: 1281
- 12a Shin C.-g, Okumura K, Ito A, Nakamura Y. Chem. Lett. 1994; 1301
- 12b Ayida BK, Simonsen KB, Vourloumis D, Hermann T. Bioorg. Med. Chem. Lett. 2005; 15: 2457
- 13 Bourland TC, Carter RG, Yokochi AF. T. Org. Biomol. Chem. 2004; 2: 1315
- 14 Zaid F, El Hajji S, El Hallaoui A, Elachqar A, Kerbal A, Roumestant ML, Viallefont P. Prep. Biochem. Biotechnol. 1998; 28: 137
- 15 Busacca CA, Grossbach D, Campbell SJ, Dong Y, Eriksson MC, Harris RE, Jones P.-J,
Kim J.-Y, Lorenz JC, McKellop KB, O’Brien EM, Qiu F, Simpson RD, Smith L, So RC, Spinelli
EM, Vitous J, Zavattaro C. J. Org. Chem. 2004; 69: 5187
Reference Ris Wihthout Link
First reports:
For reviews, see:
For recent examples, see:
-
References
- 1a Brunner H, Obermann U, Wimmer P. J. Organomet. Chem. 1986; 316: C1
- 1b Brunner H, Obermann U. Chem. Ber. 1989; 122: 499
- 1c Brunner H, Obermann U, Wimmer P. Organometallics 1989; 8: 821
- 1d Brunner H, Brandl P. J. Organomet. Chem. 1990; 390: C81
- 1e Brunner H, Wutz K, Doyle MP. Monatsh. Chem. 1990; 121: 755
- 2a Desimoni G, Faita G, Quadrelli P. Chem. Rev. 2003; 103: 3119
- 2b Aspinall HC, Greeves N. J. Organomet. Chem. 2002; 647: 151
- 2c Johnson JS, Evans DA. Acc. Chem. Res. 2000; 33: 325
- 2d Nishiyama H, Ito J.-i. Chem. Commun. 2010; 46: 203
- 2e Ito J.-i, Nishiyama H. Synlett 2012; 23: 509
- 2f Ward BD, Gade L. Chem. Commun. 2012; 48: 10587
- 3a Ingalls EL, Sibbald PA, Kaminsky W, Michael FE. J. Am. Chem. Soc. 2013; 135: 8854
- 3b De Crisci AG, Chung K, Oliver AG, Solis-Ibarra D, Waymouth RM. Organometallics 2013; 32: 2257
- 3c Narayama A, Shibatomi K, Soga Y, Muto T, Iwasa S. Synlett 2013; 24: 375
- 3d Holder JC, Marziale AN, Gatti M, Mao B, Stoltz BM. Chem. Eur. J. 2013; 19: 74
- 3e Aguado-Ullate S, Urbano-Cuadrado M, Villalba I, Pires E, Garcia JI, Bo C, Carbo JJ. Chem. Eur. J. 2012; 18: 14026
- 3f Yang G, Shen C, Zhang W. Angew. Chem. Int. Ed. 2012; 51: 9141; Angew. Chem. 2012, 124, 9275
- 3g Malkov AV, Stewart-Liddon AJ. P, McGeoch GD, Ramirez-Lopez P, Kocovsky P. Org. Biomol. Chem. 2012; 10: 4864
- 4 Christoffers J, Mann A, Pickardt J. Tetrahedron 1999; 55: 5377
- 5 Maciagiewicz IM, Fang F, Roberts DA, Zhou S, Hines JV, Bergmeier SC. Synthesis 2012; 44: 551
- 6 Griffith JA, Rowlands GJ. Synthesis 2005; 3446
- 7 Chang Y.-w, Yang J.-j, Dang J.-n, Xue Y.-x. Synlett 2007; 2283
- 8 Cook GR, Shanker PS, Peterson SL. Org. Lett. 1999; 1: 615
Reference Ris Wihthout Link
- 9a Bhor S, Anilkumar G, Tse MK, Klawonn M, Döbler C, Bitterlich B, Grotevendt A, Beller M. Org. Lett. 2005; 7: 3393
- 9b Tursky M, Necas D, Drabina P, Sedlak M, Kotora M. Organometallics 2006; 25: 901
- 9c Ma K, You J. Chem. Eur. J. 2007; 13: 1863
- 9d Ohara M, Nakamura S, Shibata N. Adv. Synth. Catal. 2011; 353: 3285
- 9e Hyodo K, Nakamura S, Shibata N. Angew. Chem. Int. Ed. 2012; 51: 10337 ; Angew. Chem. 2012, 124, 10483
- 9f Barakat A, Islam MS, Al Majid AM. A, Al-Othman ZA. Tetrahedron 2013; 69: 5185
- 9g Weber M, Klein JE. M. N, Miehlich B, Frey W, Peters R. Organometallics 2013; 32: 5810
- 9h For a review, see: Liu H, Du D.-M. Adv. Synth. Catal. 2009; 351: 489
- 10 Huang Y, Dalton DR, Carroll PJ. J. Org. Chem. 1997; 62: 372
- 11 Merritt EA, Bagley MC. J. Heterocycl. Chem. 2007; 44: 1281
- 12a Shin C.-g, Okumura K, Ito A, Nakamura Y. Chem. Lett. 1994; 1301
- 12b Ayida BK, Simonsen KB, Vourloumis D, Hermann T. Bioorg. Med. Chem. Lett. 2005; 15: 2457
- 13 Bourland TC, Carter RG, Yokochi AF. T. Org. Biomol. Chem. 2004; 2: 1315
- 14 Zaid F, El Hajji S, El Hallaoui A, Elachqar A, Kerbal A, Roumestant ML, Viallefont P. Prep. Biochem. Biotechnol. 1998; 28: 137
- 15 Busacca CA, Grossbach D, Campbell SJ, Dong Y, Eriksson MC, Harris RE, Jones P.-J,
Kim J.-Y, Lorenz JC, McKellop KB, O’Brien EM, Qiu F, Simpson RD, Smith L, So RC, Spinelli
EM, Vitous J, Zavattaro C. J. Org. Chem. 2004; 69: 5187
Reference Ris Wihthout Link
First reports:
For reviews, see:
For recent examples, see: