

RSS-Feed abonnieren
DOI: 10.1055/a-1164-1944
The Impact of Glucocorticoid Co-Secretion in Primary Aldosteronism on Thyroid Autoantibody Titers During the Course of Disease
Autor*innen

Abstract
Excess aldosterone is associated with the increased risk of cardio-/cerebrovascular events as well as metabolic comorbidities not only due to its hypertensive effect but also due to its proinflammatory action. Autonomous cortisol secretion (ACS) in the setting of primary aldosteronism (PA) is known to worsen cardiovascular outcome and potentially exhibit immunosuppressive effects. The aim of this study was to determine the impact of ACS status in patients with PA on kinetics of thyroid autoantibodies (anti-TPO, anti-TG) pre and post therapy initiation. Ninety-seven PA patients (43 unilateral, 54 with bilateral PA) from the database of the German Conn’s Registry were included. Anti-TPO and anti-TG levels were measured pre and 6–12 months post therapeutic intervention. Patients were assessed for ACS according to their 24- hour urinary cortisol excretion, late night salivary cortisol and low-dose dexamethasone suppression test. Abnormal test results in line with ACS were identified in 74.2% of patients with PA. Following adrenalectomy, significant increases in anti-TPO levels were observed in patients with at least one abnormal test (p = 0.049), adrenalectomized patients with at least two pathological ACS tests (p = 0.015) and adrenalectomized patients with pathologic dexamethasone suppression tests (p = 0.018). No antibody increases were observed in unilateral PA patients without ACS and in patients with bilateral PA receiving mineralocorticoid antagonist therapy (MRA). Our data are in line with an immunosuppressive effect of mild glucocorticoid excess in PA on thyroid autoantibody titers. This effect is uncovered by adrenalectomy, but not by MRA treatment.
Key words
primary aldosteronism - auto-immune disease - adrenalectomy - thyroid - cortisol secretion - auto-antibodiesIntroduction
Primary aldosteronism (PA) represents the leading cause of endocrine hypertension [1], accounting for 5–10% of all cases. Simultaneously, it is the most common endocrine form of hypertension that can be cured by surgery [2]. A large body of evidence exists implicating the increased risk of cardio- and cerebrovascular events as well as metabolic comorbidities in patients with aldosterone excess compared to patients with essential hypertension [3] [4] [5]. Interestingly, studies have suggested that the above mentioned complications are not only due to the hypertensive effect of excessive aldosterone but are also mediated by its proinflammatory action [6]. Recently, emphasis has been made on the effects of glucocorticoid co-secretion in the setting of PA revealing a higher incidence of metabolic syndrome and type 2 diabetes mellitus due to impaired insulin secretion [7] and insulin-sensitivity [8] further worsening the cardiovascular and metabolic burden in PA. Recent studies have investigated the implication of aldosterone in the development and/or progression of autoimmune disorders, as aldosterone is associated with the activation of circulating immune cells, reduced by mineralocorticoid receptor antagonists (MRA) [9]. Autoimmune thyroiditis also known as Hashimoto’s thyroiditis represents one of the most common disorders in the general population, especially in women, with the incidence estimated at 3.5 cases/1000 women per year [10] [11]. In the setting of autoimmunity, Hashimoto’s thyroiditis is characterized by the presence of specific autoantibodies (most specifically anti-TPO) [10] and by a lymphocytic infiltration [10] [11]. In this study, we investigated the kinetics of anti-thyroid peroxidase (anti-TPO) and thyroglobulin antibody (anti-TG) in patients with primary aldosteronism and evaluated the extent of concomitant autonomous cortisol secretion (ACS) on said antibodies pre- and post-therapy initiation.
Patients and Methods
Description of the patient cohort and diagnostic workup
The study population consisted of 97 patients who were recruited at the Munich center of the German Conn’s Registry. The German Conn’s Registry is a multicenter-registry that investigates therapy, comorbidities and the long-term outcome in PA patients throughout Germany since 2008 [12]. The investigated cohort was studied between January 2014 and February 2019. The diagnosis of PA was established as stated in the Endocrine Society Practice Guidelines [2] . Diagnosis was established initially during screening by an elevated blood pressure (BP) and an elevated plasma aldosterone/renin ratio (ARR) (cutoff: 12.0 ng/U sitting position) [13] followed by an abnormal confirmatory test (saline infusion and/or captopril challenge test). Before conducting these tests (and complete testing for hypercortisolism) interfering antihypertensive medication was changed whenever possible (>90% of patients). ACE inhibitors, beta blockers, central-alpha agonists, angiotensin receptor blockers (ARB’s) were withdrawn for at least one week, and mineralocorticoid receptor antagonists (MRA) for at least four weeks prior testing. The diagnosis was then made on the basis of all clinical and laboratory findings. All patients in this study underwent adrenal venous sampling without cosyntropin stimulation as well as some form of adrenal imaging (computed tomography scanning or MRI) for subtype differentiation between unilateral and bilateral disease. The majority of patients (>80%) had simultaneous bilateral adrenal venous sampling using a selectivity index of ≥2.0 and a lateralization index of ≥4.0 [14] [15] [16].
All patients underwent complete testing for hypercortisolism, including 1 mg dexamethasone suppression test (DST), a 24-hour urinary free cortisol determination (UFC) and late night salivary cortisol (LSC) measurement at baseline visit. Furthermore, anti-TPO and anti-TG as well as thyroid function (TSH, fT3 and fT4) were tested at baseline and during follow up visits. All patients underwent thyroid ultrasound at baseline visit.
Patients with missing data for aldosterone, renin, serum potassium, anti-TPO, anti-TG, blood pressure, DST, 24-hour urinary cortisol or LSC were excluded from this study. Other exclusion criteria were patients with preexisting hyperthyroidism or biochemical overt hyperthyroidism at any of the visits, patients taking thyreostatic medications, patients with history of thyroid cancer and patients who had undergone thyroid surgery. Unilateral adrenalectomy (ADX) was offered to all patients with unilateral PA. Patients with unilateral PA who did not opt for ADX were excluded from this study. All patients with bilateral PA were treated with MRA using spironolactone with a starting dose of 25–50 mg per day in the majority of the cases. All patients had a follow up visit (V2) one year after being treated with either MRA or ADX.
Definitions and laboratory measurements
Measurement of serum cortisol was conducted using an immunochemiluminescence assay (Liaison; DiaSorin, Saluggia, Italy). Within and between intra-assay and interassay variability was <10%, with a lower limit of quantification at 0.2 μg/dl as previously described [13] [14] [17]. Urinary cortisol measurement was performed with chemiluminescence immunoassay (ADVIA Centaur, Siemens) with within- and between-assay coefficients variations below 7%. Before 01.06.2016, salivary cortisol was measured by a luminescence immunoassay (Cortisol LIA, IBL, Hamburg, Germany) with within- and between-assay coefficients variations below 9% and 6%. Since 01.06.2016, salivary cortisol was measured by an automated chemiluminescence immunoassay (IDS-iSYS CLIA, Immunodiagnostic Systems, Boldon, UK) with within- and between-assay coefficients variations below 8% and 12%, respectively. Serum aldosterone measurement was performed with chemiluminescence immunoassay (LIAISON, CLIA, DiaSorin). Anti-TPO and anti-TG titers were obtained at baseline and at follow up visit (reference values 1–16 IU/ml and 5–100 IU/ml, respectively) and measurement was performed with chemiluminescence immunoassay (Liaison; CLIA, DiaSorin).
ACS was assumed when DST, LSC or UFC were elevated [Reference values: >1.8 μg/dl, >1.5 ng/ml, >150 μg/l (pre 2015) or >83 μg/l (post 2015), respectively]. Blood pressure was measured 3 times on each arm after at least 5 minutes of resting with standard sphygmomanometers. Body mass index (BMI) was calculated as body weight (kg) per heights (m2).
Statistical analysis
In a priori power analysis, we calculated that a total sample size of 72 would be sufficient to identify significant differences between groups, assuming a medium effect (0.6), a power of 1–β = 0.80 and a type I error of α = 0.05, with 36 subjects having unilateral PA and 36 subjects having bilateral idiopathic hyperplasia (IHA). Statistical analysis was done using standard statistical software (SPSS 25, IBM, Chicago, IL, USA). All values are expressed as median, 25th and 75th percentile if not mentioned otherwise. Data between normal and pathological subgroups were compared using Mann–Whitney U-test. To study the impact of increasing levels of ACS, patients were grouped according the number of pathological tests into the following groups: 0–1 pathological tests, or 2 and more pathological tests. Since only 5 subjects had 3 pathological tests, we considered this group to be borderline small and combined it with the group of patients with 2 pathological tests.
Within-group changes from baseline to follow-up were calculated by Wilcoxon signed-rank test. Stepwise multiple regression analysis was performed for multivariate analysis. Differences were considered statistically significant when p ≤0.05. Categorical variables are displayed as percentage or numbers. Since urinary cortisol reference ranges differed depending on date of visit (>150 µg/l until 2015, >83 μg/l post 2015) we calculated the upper limit of normal to form a general collective.
To evaluate possible significant differences in antibody kinetics between two patient collectives (visit 1 vs. follow up visit), the delta and subsequently absolute proportion of the anti-TPO and anti-TG between baseline and follow up was calculated. A linear regression was then performed.
Results
Of our 97 investigated PA patients, 43 underwent ADX whereas 54 patients have been diagnosed with IHA and have been treated with MRA. Of these patients ACS was identified in 74.2% (n = 72, 37 with one pathological test, 28 with two pathological tests, 7 with three pathological tests). We found no significant differences in anti-TPO (and anti-TG) titers at baseline between ACS and non ACS patients ([Fig. 1] and [Table 1]). Fourteen patients (14.4%, 5 in the ADX group, 9 in the MRA group) showed either elevated anti-TPO and/or anti-TG at baseline visit. All patients received follow up visits one year after therapy initiation and characteristics are shown in [Table 2]. When comparing the prevalence of ACS according to subtypes, the presence of ACS was evident in both subgroups. At least one pathological test indicating ACS was present in 84% of unilateral PA patients and in 67% of IHA patients ([Table 2]). Thirteen of the analyzed patients (6 ADX, 7 MRA) were taking thyroid hormone supplementation either at baseline or during the follow up visit. All dosages remained the same post therapy initiation. Eleven Patients (6 ADX, 5 MRA) had known preexisting Hashimoto thyroiditis, based on typical ultrasound findings, antibody titers and/or hypothyroidism.

|
Characteristics |
ACS |
No ACS |
p |
|---|---|---|---|
|
n (%) |
72 (74.2) |
25 (25.8) |
0.000 |
|
Male n (%) |
49 (68.1) |
12 (48) |
0.010 |
|
Age (years) |
57.7±10 |
50±10 |
0.000 |
|
BMI (kg/m2) |
27.6± 5.89 (27.6; 24.2–32.1) |
26.7±4.46 (26.7; 23.9–29.7) |
0.276 |
|
Systolic BP (mmHg) |
149.5± 17.9 (149.5; 135-160) |
147±23.7 (147; 132–160) |
0.496 |
|
Diastolic BP (mmHg) |
87±11.6 (87; 82–98) |
93±14.4 (93; 81–101) |
0.455 |
|
Aldosterone pg/ml |
149±233 (149; 102–249) |
162±117.4 (162; 114–270) |
0.509 |
|
PRC mU/l |
3.6±4.8 (3.6; 2–7.1) |
3.2±4.2 (3.2; 2–5.3) |
0.863 |
|
ARR |
37.3±83.3 (37.3; 22.5–70.1) |
49.10±63.5 (49.10; 21.5–81.1) |
0.421 |
|
Potassium (mmol/l) |
3.6±0.5 (3.6; 3.4–3.9) |
3.7±0.3 (3.7; 3.5–3.8) |
0.505 |
|
Anti TPO (IU/ml) |
2.6±107 (2.6; 1.4–5.7) |
3.2±12.1 (3.2; 1.6–4.7) |
0.928 |
|
Anti TG (IU/ml) |
6.3±40.3 (6.3; 5–10) |
7±33.5 (7; 5.4–11.2) |
0.218 |
|
TSH (IU/ml) |
1.7±0.9 (1.7; 1.1–2.2) |
1.8±5.6 (1.8; 1.3–2.2) |
0.544 |
|
HbA1c (mmol/mol) |
34±7 (34; 31–39) |
36±7 (36; 32–37) |
0.769 |
|
Glucose (mg/dl) |
100±18.8 (100; 90.1–108) |
95±20.6 (95; 91–105) |
0.281 |
|
Total cholesterol (mg/dl) |
190±36.6 (190; 168.5–220) |
182±29.7 (182; 158–198) |
0.073 |
|
LDL cholesterol (mg/dl) |
117±39.1 (117; 94–148.5) |
112±31.7 (112; 77–126) |
0.150 |
|
HDL cholesterol (mg/dl) |
55±15.8 (55; 45–66) |
51±18.7 (51; 43–64) |
0.629 |
|
Triglycerides (mg/dl) |
95.5±101.2 (95.5; 69.5–133.5) |
105±45.5 (105; 58–134) |
0.683 |
|
Basal cortisol (µg/dl) |
13.5 ± 5.5 (13.5; 9.8–17.4) |
12.1±5.5 (12.1; 7.8–17) |
0.362 |
|
Cortisol after 1 mg DST (μg/dl) |
1.5±2.5 (1.5; 1.2–1.8) |
1.2±0.5 (1.2; 1–1.4) |
0.009 |
|
Late night salivary cortisol (ng/ml) |
1.6±1 (1.6; 0.9–2.2) |
0.9±0.3 (0.9; 0.7–1) |
0.000 |
|
Urinary free cortisol (μg/24 h) upper limit of normal |
1.3±3 (1.3; 1.1–2.1) |
0.8±0.3 (0.8; 0.5–0,9) |
0.000 |
Data are displayed as mean±SD for normally distributed continuous data, and as median; 25th and 75th continuous variables without normal distribution. BMI: Body mass index; PRC: Plasma renin concentration; ARR: Aldosterone/renin ratio; Anti-TPO: Anti-thyroid peroxidase; anti-TG: Anti-thyroglobulin; BP: Blood pressure; DST: Dexamethasone suppression test; LSC: Late night salivary cortisol; UFC: Urinary-free cortisol.
|
Variables |
ADX |
MRA |
||||
|---|---|---|---|---|---|---|
|
Baseline (V1) |
Follow up (V2) |
p |
Baseline (V1) |
Follow up (V2) |
p |
|
|
ACS/no ACS n (%) |
36/7 (83.7/16.3) |
– |
36/18 (66.7/33.3) |
– |
||
|
Male n (%) |
29 (67.4) |
– |
32 (59.3) |
– |
||
|
Systolic BP (mmHg) |
149 ± 18.9 (149; 133–158) |
136±17.7 (136; 124–141) |
0.003 |
149±19.9 (149; 133–160) |
132±14.8 (132; 122–143) |
0.000 |
|
Diastolic BP (mmHg) |
87±11.6 (87; 81–98) |
86±11.5 (86; 81–98) |
0.814 |
91±13 (91; 83–100) |
87±9.7 (87; 82–93) |
0.095 |
|
Potassium (mmol/l) |
3.5±0.6 (3.5; 3.3–3.7) |
4.3±7.2 (4.3; 4.1–4.5) |
0.000 |
3.75±0.3 (3.75; 3.6–3.9) |
4.3±0.4 (4.3; 4.1–4.6) |
0.000 |
|
Aldosterone (pg/ml) |
218±281.1 (218; 121–353) |
65±49.9 (65; 40–114) |
0.000 |
129.5±93.7 (129.5; 99–186) |
266.5±134.6 (266.5; 173–334) |
0.000 |
|
PRC (mU/l) |
4.4 ±5.5 (4.4; 2–8.5) |
11.9±38.4 (11.9; 7.4–21.3) |
0.000 |
3.3±3.8 (3.3; 2–5.3) |
13.2±129.5 (13.2; 7–32) |
0.000 |
|
ARR |
47.8±102.7 (48; 23.5–165.2) |
5.2±21 (5.2; 2.3–13.7) |
0.000 |
38.2±44.2 (38.2; 20.8–62) |
16.2 ±25.6 (16.2; 9.8–29.7) |
0.000 |
|
Anti TPO (IU/ml) |
2.6±10.7 (2.6; 1.6–4.7) |
2.8±22 (2.8; 1.6–6.8) |
0.181 |
2.85±123.2 (2.85; 1.4–5.6) |
3.45±87.2 (3.45; 1.1–7.8) |
0.714 |
|
Anti TG (IU/ml) |
6.4±45 (6.4; 5–11.4) |
7.4±48.2 (7.4; 6–13.5) |
0.433 |
6.7±32.9 (6.7; 5–10.3) |
7.2±43.9 (7.2; 5.8–12) |
0.487 |
|
TSH (IU/mL) |
1.7±1 (1.7; 0.9–2) |
1.62±1 (1.62; 1.3–2.3) |
0.125 |
1.8±3.9 (1.8; 1.3–2.2) |
2±1 (2; 1.2–2.6) |
0.267 |
|
HbA1c (mmol/mol) |
36±8 (36; 31–40) |
38±9 (38; 33–42) |
0.000 |
33±7 (33; 31–37) |
36±8 (36; 33–40) |
0.000 |
|
Glucose (mg/dl) |
97±22.5 (97; 90–113) |
99±21 (99; 91–114) |
0.735 |
99±16.3 (99; 91–106) |
98±22.5 (98, 93–109) |
0.012 |
|
Total cholesterol (mg/dl) |
185±39.1 (185; 168–211) |
192±43.2 (192; 54–219) |
0.432 |
190.5±32.2 (190.5; 163–212) |
191±37.6 (191; 164–212) |
0.703 |
|
LDL cholesterol (mg/dl) |
120±40.6 (120; 90–140) |
110±39.8 (110; 86–136) |
0.717 |
115±35.6 (115; 91–141) |
111±40.5 (111; 90–138) |
0.275 |
|
HDL cholesterol (mg/dl) |
55±15.1 (55; 45–64) |
50±16.2 (50; 41–65) |
0.000 |
51 ±17.6 (51; 44–66) |
51±17.4 (51; 43–64) |
0.035 |
|
Triglycerides (mg/dl) |
88±46.7 (88; 66–130) |
114±96.9 (114; 85–168) |
0.000 |
105.5±113.2 (105.5; 70–134) |
127±128.9 (127; 97–159) |
0.001 |
Data are displayed as mean±SD for normally distributed continuous data, and as median; 25th and 75th continuous variables without normal distribution. ADX: Adrenalectomy; no ACS: No pathologic cortisol test regarding hypercortisolism; BMI: Body mass index; PRC: Plasma renin concentration; ARR: Aldosterone/renin ratio; Anti-TPO: Anti-thyroid peroxidase; anti-TG: anti-thyroglobulin; BP: Blood pressure; DST: Dexamethasone suppression test; LSC: Late night salivary cortisol; UFC: Urinary-free cortisol.
To further explore the effects of ACS, we analyzed the course of thyroid antibody titers in patients who underwent ADX. Taking into account all adrenalectomized patients regardless of ACS status, no significant differences were noted with respect to anti-TPO and anti-TG titers (p = 0.181 and 0.433, respectively) whereas adrenalectomized ACS patients showed an increase of anti-TPO titers of borderline significance ([Fig. 2]).
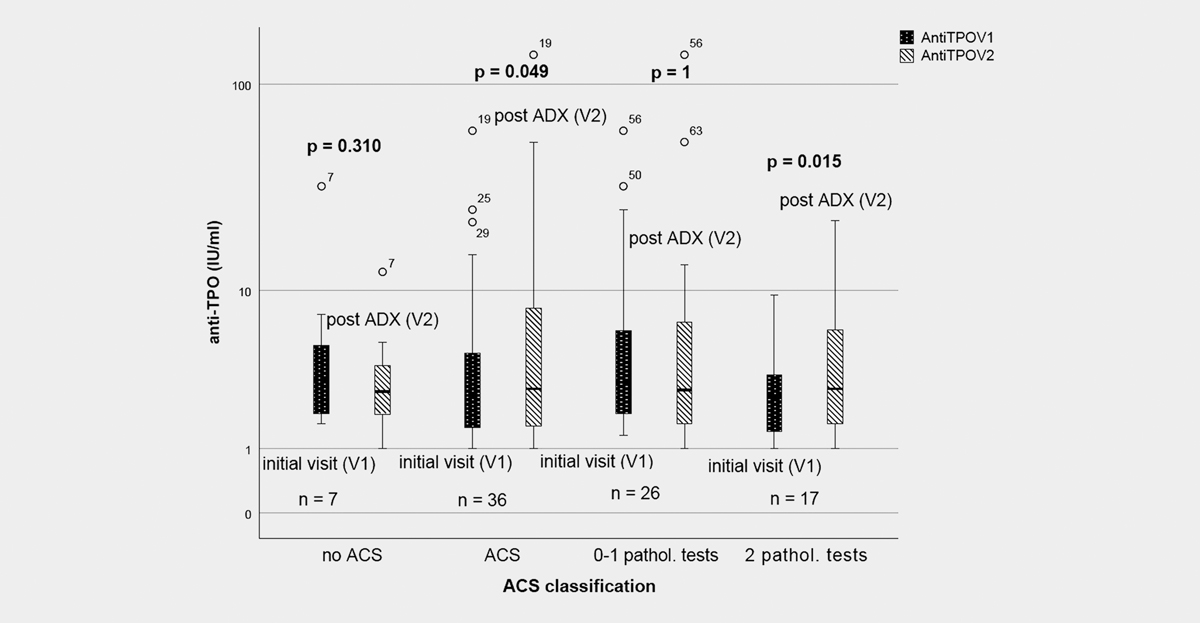
Adrenalectomized patients with no or one pathological ACS test (n = 26) showed no significant increase in antibody titers. In contrast, patients with two or more pathological screening tests (n = 17) had significantly higher anti-TPO levels (p = 0.015) after ADX ([Fig. 2]). Anti-TG levels remained unchanged following ADX (p = 0.726).
In patients with at least two pathological screening tests, compared to non ACS patients, the impact of ACS subtype on anti-TPO titers remained significant when adjusted for age (p = 0.010) or age and gender (p = 0.021). When further adjusting for BMI, systolic BP and ARR, ACS status stayed significant (p = 0.038), further underlining its influence on anti-TPO levels. ACS subtype was not associated with anti-TG titers (p = 0.448) even when adjusting for age and gender.
Patients with IHA receiving MRA treatment did not show differences in thyroid antibody titers during follow-up, irrespective of ACS status.
Finally, we analyzed the effect of ACS in terms of the DST results, on our primary outcome parameter, the anti-TPO antibody increase. Sixteen (8 unilateral, 8 IHA patients) of 97 patients (16.5%) were found to have a pathological DST response. PA patients who have undergone ADX showed a significant anti-TPO titer increase (p = 0.018), whereas patients treated with MRA displayed no such rise during follow-up.
We also analyzed patients that initially displayed elevated anti-TPO and/or anti-TG levels. Of these 14 patients (4 unilateral, 10 IHA patients), 5 had been previously diagnosed with Hashimoto’s thyroiditis and were taking l-thyroxine during their initial visit. These patients showed higher absolute antibody levels and TSH compared to the rest of the patients (3.69 vs. 1.79 μU/ml). Patients with initially elevated antibodies showed a higher BMI pre- and post-therapy (31 kg/m2 at V1 vs. 26 kg/m2 in PA patients with normal thyroid antibody titers at V1), and a significantly higher ARR at follow up (16.2 vs. 11.4, p = 0.006).
Unfortunately, we only have limited data regarding hypercortisolism at follow up, which is why an evaluation regarding the resolution of ACS post ADX or during MRA therapy could not be sufficiently made in this study. A follow up 24-hour urinary free cortisol was available in 8 patients with ACS who had undergone ADX, with the majority of these patients showing a decline in their 24-hour urinary cortisol excretion (mean 227 μg/dl before ADX vs. 120 μg/dl after 1 year).
Discussion
Studies investigating the interaction between ACS in the setting of primary aldosteronism and its effect on thyroid autoantibodies are scarce. Aldosterone has been shown to promote an inflammatory state that is enhanced by T cell immunity, therefore establishing its role in the immune system. Studies have shown that the capacity of dendritic cells to activate CD8+T cells and induce Th17 polarization of CD4+ T cells, known to occur in many autoimmune diseases, can directly be increased by aldosterone [18]. Aldosterone encourages inflammation characterized by vascular infiltration of immune cells, proinflammatory cytokine production (e. g., TNF-α), and reactive oxidative stress. This may underline the essential role of aldosterone and its contribution to the development and progression of autoimmunity [18] [19] [20] [21] . Fittingly, Herrada and Shi et al. [18] [20] have found an increased expression of Th17-related factors in autoimmune thyroiditis (AIT).
Bendtzen et al. [22] demonstrated that the mineralocorticoid receptor antagonist spironolactone is able to reduce tumor necrosis factor-α and interferon-γ production in patients with rheumatoid and juvenile idiopathic arthritis, thus reducing inflammation.
The exact pathophysiology of ACS in IHA is unknown and remains closely investigated until this day. Diffuse or nodular hyperplasia of the zona glomerulosa is considered the traditional source of excess aldosterone in these patients. While diffuse hyperplasia of the zona glomerulosa cells in IHA adrenals have commonly been reported [23] [24] [25] [26] [27], other studies describe the presence of hyperplasia of zona glomerulosa and zona fasciculate like cells which may serve as a proposed mechanism of ACS in IHA [24] [25] [28] [29] [30] [31] [32]. These findings have been referred to as primary cortical hyperplasia.
Several studies have found that titers of angiotensin-II type-1 receptor autoantibodies (AT1RAA) were elevated in PA patients, indicating a potential pathophysiologic role for agonistic autoantibodies in PA [33] [34]. It has been suggested that approximately 60% of PA patients possess active autoantibodies against AT1R [35]. As a result, the AT1R may be activated by the production of these antibodies, which may contribute to the development of PA in some patients [36]. Er et al. [37] hypothesized that this process raises the possibility of immune dysregulation in PA, which in turn carries an increased risk of developing concomitant autoimmune disease. Interestingly, Piazza et al. [38] investigated the kinetics of AT1R titers in APA patients and found that these patients showed elevated levels in comparison to IHA patients. Furthermore, the raised autoantibody titers in the APA patient cohort did not change 1 month after unilateral adrenalectomy. This finding, together with our own data, raise the possibility that mild functional hypercortisolism (i. e., ACTH triggered) rather than autonomous secretion of cortisol by the APA may be the responsible mechanism, which would implicate less effects on the immune system. This remains the subject for further investigation.
It has been proven that the prevalence of thyroid morphological alterations in PA patients is high compared to the general population [39] . Our study further explored the interactions between anti-TPO and anti-TG levels in the setting of primary aldosteronism with and without ACS. In a study conducted by Armanini et al. [40], a statistically significant difference in the prevalence of thyroid abnormalities between PA and healthy controls (60% vs. 27%, p <0.0001) has been demonstrated. Turchi et al. also studied the relationship between aldosterone and thyroid disease among 188 patients with PA and 96 matched essential hypertensives. Here the prevalence of thyroid dysfunction was similar in PA and essential hypertension (EH) (15% and 19%, respectively) [39].
Our study shows that patients with unilateral disease and significant cortisol co-secretion, especially those with at least two pathological cortisol tests, demonstrated an increase in TPO antibody titers after surgery compared to patients with no ACS. This effect stayed fairly robust even when adjusted for age and gender. Along the same line, adrenalectomized patients with ACS and a pathological DST response showed a significant increase in anti-TPO titers after surgery compared to ACS patients with IHA and a pathologic DST response. Currently, there are several case reports of exacerbation of Hashimoto’s thyroiditis or Graves’ disease following partial hypophysectomy or adrenalectomy in patients with Cushing’s disease [41] [42] [43] [44] .
Other studies have reported the onset of rheumatoid arthritis [45] [46] and systemic lupus erythematosus [47] after successful surgical treatment of Cushing’ disease. Immunosuppressive effects exhibited by cortisol are well known, which inhibits most aspects of the immune response, including proliferation of lymphocytes. By means of reducing T cell proliferation, glucocorticoids are known to suppress autoimmune reactions [48]. It is due to these effects that they have been used to treat patients with Hashimoto’s thyroiditis, to decrease anti-TPO and anti-TG antibodies and ultimately normalize thyroid function [49]. Corollary, it has been suggested that patients with Cushing’s syndrome and concomitant thyroid autoimmune disease may be protected by the exhibited immunosuppressive effects of hypercortisolism [43]. By treating the disease (removal of adrenal or pituitary adenomas), the autoimmune process as a result of restoration of normal pituitary-adrenal function, may be intensified and overt thyroid dysfunction is likely to develop. A study conducted by Niepomniszcze et al. [50] in patients with Cushing’s disease and autoimmune thyroid disease showed that the prevalence of positive anti thyroid antibodies during active disease (27%) increased drastically (87%) after hypercortisolism resolved. These findings corroborate our results. Colao et al. [51] also documented an increased prevalence of anti-TG and anti-TPO positive titers in patients following remission of Cushing’s disease as compared during active disease (20% and 40% pre- and post-successful pituitary surgery).
Er et al. further analyzed the risk of new onset autoimmune disease in PA patients. This study mirrored our results by showing a higher incidence of each autoimmune disease (incl. inflammatory conditions) in patients with PA compared to matched EH controls. Fittingly, patients who underwent either ADX or MRA treatment still showed an increased incident risk of developing new onset autoimmune disease [37].
With that being said, no significant differences between TSH levels were found in ACS versus non-ACS patients in our study, suggesting that the relatively small titer changes observed in our patients are not associated with clinical AIT. A weakness of our study is the lack of a gender and age-matched normotensive control group. Moreover, antibody values from baseline visits were often missing, causing us to eliminate many potential patients from further analysis, therefore leading to a smaller sample size.
Another limitation of this study was that an evaluation regarding the normalization of ACS after surgery or during MRA therapy could not sufficiently be made due to limited hypercortisolism testing available during follow up visits.
All in all, overall changes in antibody kinetics are slight, therefore not exhibiting a significant clinical effect, although from a pathophysiological standpoint these findings are fitting to the immunosuppressive agency of cortisol. A key strength of the present study was that we conducted a prospective analysis of patients within the conditions of a standardized registry including the usage of same assays during initial visit and follow up. As far as we know, this is the first study investigating the effect of ACS in the setting of PA on thyroid autoantibodies pre and post therapy initiation.
Conclusion
This study investigated the impact of glucocorticoid co-secretion in PA on thyroid autoantibodies during the course of disease. In conclusion, unilateral ADX in patients with PA and significant cortisol co-secretion is associated with anti-TPO antibody titer increases, a finding, which is not observed in MRA treated patients.
Conflict of Interest
The authors declare that they have no conflict of interest.
-
References
- 1 Rossi GP, Bernini G, Caliumi C. et al. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients. J Am Coll Cardiol 2006; 48: 2293-2300
- 2 Funder JW, Carey RM, Mantero F. et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016; 101: 1889-1916
- 3 Born-Frontsberg E, Reincke M, Rump LC. et al. Cardiovascular and cerebrovascular comorbidities of hypokalemic and normokalemic primary aldosteronism: Results of the German Conn’s Registry. J Clin Endocrinol Metab 2009; 94: 1125-1130
- 4 Milliez P, Girerd X, Plouin PF. et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45: 1243-1248
- 5 Quinkler M, Born-Frontsberg E, Fourkiotis VG. Comorbidities in primary aldosteronism. Horm Metab Res 2010; 42: 429-434
- 6 Pitt B. Effect of aldosterone blockade in patients with systolic left ventricular dysfunction: Implications of the RALES and EPHESUS studies. Mol Cell Endocrinol 2004; 217: 53-58
- 7 Fischer E, Adolf C, Pallauf A. et al. Aldosterone excess impairs first phase insulin secretion in primary aldosteronism. J Clin Endocrinol Metab 2013; 98: 2513-2520
- 8 Colussi G, Catena C, Lapenna R. et al. Insulin resistance and hyperinsulinemia are related to plasma aldosterone levels in hypertensive patients. Diabetes Care 2007; 30: 2349-2354
- 9 Ahokas RA, Warrington KJ, Gerling IC. et al. Aldosteronism and peripheral blood mononuclear cell activation: A neuroendocrine-immune interface. Circ Res 2003; 93: e124-e135
- 10 Caturegli P, Kimura H, Rocchi R. et al. Autoimmune thyroid diseases. Curr Opin Rheumatol 2007; 19: 44-48
- 11 Lorini R, Gastaldi R, Traggiai C. et al. Hashimoto’s Thyroiditis. Pediatr Endocrinol Rev 2003; 1 (Suppl. 02) 205-211 discussion 211.
- 12 Schirpenbach C, Segmiller F, Diederich S. et al. The diagnosis and treatment of primary hyperaldosteronism in Germany: Results on 555 patients from the German Conn Registry. Dtsch Arztebl Int 2009; 106: 305-311
- 13 Schilbach K, Junnila RK, Bidlingmaier M. Aldosterone to renin ratio as screening tool in primary aldosteronism. Exp Clin Endocrinol Diabetes 2019; 127: 84-92
- 14 Betz MJ, Degenhart C, Fischer E. et al. Adrenal vein sampling using rapid cortisol assays in primary aldosteronism is useful in centers with low success rates. Eur J Endocrinol 2011; 165: 301-306
- 15 Ladurner R, Sommerey S, Buechner S. et al. Accuracy of adrenal imaging and adrenal venous sampling in diagnosing unilateral primary aldosteronism. Eur J Clin Invest 2017; 47: 372-377
- 16 Riester A, Fischer E, Degenhart C. et al. Age below 40 or a recently proposed clinical prediction score cannot bypass adrenal venous sampling in primary aldosteronism. J Clin Endocrinol Metab 2014; 99: E1035-E1039
- 17 Heinrich DA, Adolf C, Holler F. et al. Adrenal insufficiency after unilateral adrenalectomy in primary aldosteronism: Long-term outcome and clinical impact. J Clin Endocrinol Metab 2019; 104: 5658-5664
- 18 Herrada AA, Contreras FJ, Marini NP. et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol 2010; 184: 191-202
- 19 Herrada AA, Campino C, Amador CA. et al. Aldosterone as a modulator of immunity: Implications in the organ damage. J Hypertens 2011; 29: 1684-1692
- 20 Shi Y, Wang H, Su Z. et al. Differentiation imbalance of Th1/Th17 in peripheral blood mononuclear cells might contribute to pathogenesis of Hashimotoʼs thyroiditis. Scand J Immunol 2010; 72: 250-255
- 21 Yoshimoto T, Hirata Y. Aldosterone as a cardiovascular risk hormone. Endocr J 2007; 54: 359-370
- 22 Bendtzen K, Hansen PR, Rieneck K. et al. Spironolactone inhibits production of proinflammatory cytokines, including tumour necrosis factor-alpha and interferon-gamma, and has potential in the treatment of arthritis. Clin Exp Immunol 2003; 134: 151-158
- 23 Munro Neville A. MJOH. Hyperaldosteronism and Related Syndromes of Mineralocorticoid Excess. Springer; London: 1982: 202-241
- 24 Biglieri EG, Schambelan M, Slaton PE. et al. The intercurrent hypertension of primary aldosteronism. Circ Res 1970; 27: 195-202
- 25 Enberg U, Volpe C, Hoog A. et al. Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mRNA expression of the gene CYP11B2. Eur J Endocrinol 2004; 151: 73-85
- 26 Nakamura Y, Felizola SJ, Satoh F. et al. Dissecting the molecular pathways of primary aldosteronism. Pathol Int 2014; 64: 482-489
- 27 Omata K, Satoh F, Morimoto R. et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension 2018; 72: 874-880
- 28 Davis WW, Newsome HH, Wright LD. et al. Bilateral adrenal hyperplasia as a cause of primary aldosteronism with hypertension, hypokalemia and suppressed renin activity. Am J Med 1967; 42: 642-647
- 29 Doorenbos H, Elings HS, Van Buchem FS. Primary aldosteronism due to adrenocortical hyperplasia. Lancet 1956; 271: 335-337
- 30 Ganguly A. Primary aldosteronism. N Engl J Med 1998; 339: 1828-1834
- 31 Katz FH. Primary aldosteronism with suppressed plasma renin activity due to bilateral nodular adrenocortical hyperplasia. Ann Intern Med 1967; 67: 1035-1042
- 32 Ledingham JG, Laragh JH, Sommers SC. Secondary aldosteronism and reduced plasma renin in hypertensive disease. Trans Assoc Am Physicians 1967; 80: 168-182
- 33 Kem DC, Li H, Velarde-Miranda C. et al. Autoimmune mechanisms activating the angiotensin AT1 receptor in ‛primary’ aldosteronism. J Clin Endocrinol Metab 2014; 99: 1790-1797
- 34 Rossitto G, Regolisti G, Rossi E. et al. Elevation of angiotensin-II type-1-receptor autoantibodies titer in primary aldosteronism as a result of aldosterone-producing adenoma. Hypertension 2013; 61: 526-533
- 35 Li H, Yu X, Cicala MV. et al. Prevalence of angiotensin II type 1 receptor (AT1R)-activating autoantibodies in primary aldosteronism. J Am Soc Hypertens 2015; 9: 15-20
- 36 Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 2015; 100: 1-10
- 37 Er LK, Chen L, Tsai YC. et al. Risk of new-onset autoimmune diseases in primary aldosteronism: A nation-wide population-based study. J Hypertens 2019; 38: 745-754
- 38 Piazza M, Seccia TM, Caroccia B. et al. AT1AA (Angiotensin II Type-1 Receptor Autoantibodies): Cause or consequence of human primary aldosteronism?. Hypertension 2019; 74: 793-799
- 39 Turchi F, Ronconi V, di Tizio V. et al. Blood pressure, thyroid-stimulating hormone, and thyroid disease prevalence in primary aldosteronism and essential hypertension. Am J Hypertens 2011; 24: 1274-1279
- 40 Armanini D, Nacamulli D, Scaroni C. et al. High prevalence of thyroid ultrasonographic abnormalities in primary aldosteronism. Endocrine 2003; 22: 155-160
- 41 Cioni K, Pantaleoni M, Toschi E. et al. Exacerbation of autoimmune hypothyroidism after hemi-hypophysectomy in a patient with Cushing's disease. Minerva Endocrinol 1993; 18: 139-141
- 42 Colombo P, Passini E, Re T. et al. Autoimmune thyroid disease after successful treatment of Cushing’s syndrome. J Endocrinol Invest 1994; 17: 289-290
- 43 Takasu N, Komiya I, Nagasawa Y. et al. Exacerbation of autoimmune thyroid dysfunction after unilateral adrenalectomy in patients with Cushing’s syndrome due to an adrenocortical adenoma. N Engl J Med 1990; 322: 1708-1712
- 44 Yamakita N, Sakata S, Hayashi H. et al. Case report: Silent thyroiditis after adrenalectomy in a patient with Cushing’s syndrome. Am J Med Sci 1993; 305: 304-306
- 45 Uthman I, Senecal JL. Onset of rheumatoid arthritis after surgical treatment of Cushing’s disease. J Rheumatol 1995; 22: 1964-1966
- 46 Yakushiji F, Kita M, Hiroi N. et al. Exacerbation of rheumatoid arthritis after removal of adrenal adenoma in Cushing’s syndrome. Endocr J 1995; 42: 219-223
- 47 Noguchi Y, Tamai H, Fujisawa K. et al. Systemic lupus erythematosus after pituitary adenomectomy in a patient with Cushing’s disease. Clin Endocrinol (Oxf) 1998; 48: 670-672
- 48 McGregor AM. Immunoendocrine interactions and autoimmunity. N Engl J Med 1990; 322: 1739-1741
- 49 Murray IP. The effect of prednisolone on Hashimoto’s thyroiditis. Scott Med J 1958; 3: 341-345
- 50 Niepomniszcze H, Pitoia F, Katz SB. et al. Primary thyroid disorders in endogenous Cushing's syndrome. Eur J Endocrinol 2002; 147: 305-311
- 51 Colao A, Pivonello R, Faggiano A. et al. Increased prevalence of thyroid autoimmunity in patients successfully treated for Cushing's disease. Clin Endocrinol (Oxf) 2000; 53: 13-19
Correspondence
Publikationsverlauf
Eingereicht: 20. Dezember 2019
Angenommen: 20. April 2020
Artikel online veröffentlicht:
13. Mai 2020
© 2020. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Rossi GP, Bernini G, Caliumi C. et al. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients. J Am Coll Cardiol 2006; 48: 2293-2300
- 2 Funder JW, Carey RM, Mantero F. et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016; 101: 1889-1916
- 3 Born-Frontsberg E, Reincke M, Rump LC. et al. Cardiovascular and cerebrovascular comorbidities of hypokalemic and normokalemic primary aldosteronism: Results of the German Conn’s Registry. J Clin Endocrinol Metab 2009; 94: 1125-1130
- 4 Milliez P, Girerd X, Plouin PF. et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005; 45: 1243-1248
- 5 Quinkler M, Born-Frontsberg E, Fourkiotis VG. Comorbidities in primary aldosteronism. Horm Metab Res 2010; 42: 429-434
- 6 Pitt B. Effect of aldosterone blockade in patients with systolic left ventricular dysfunction: Implications of the RALES and EPHESUS studies. Mol Cell Endocrinol 2004; 217: 53-58
- 7 Fischer E, Adolf C, Pallauf A. et al. Aldosterone excess impairs first phase insulin secretion in primary aldosteronism. J Clin Endocrinol Metab 2013; 98: 2513-2520
- 8 Colussi G, Catena C, Lapenna R. et al. Insulin resistance and hyperinsulinemia are related to plasma aldosterone levels in hypertensive patients. Diabetes Care 2007; 30: 2349-2354
- 9 Ahokas RA, Warrington KJ, Gerling IC. et al. Aldosteronism and peripheral blood mononuclear cell activation: A neuroendocrine-immune interface. Circ Res 2003; 93: e124-e135
- 10 Caturegli P, Kimura H, Rocchi R. et al. Autoimmune thyroid diseases. Curr Opin Rheumatol 2007; 19: 44-48
- 11 Lorini R, Gastaldi R, Traggiai C. et al. Hashimoto’s Thyroiditis. Pediatr Endocrinol Rev 2003; 1 (Suppl. 02) 205-211 discussion 211.
- 12 Schirpenbach C, Segmiller F, Diederich S. et al. The diagnosis and treatment of primary hyperaldosteronism in Germany: Results on 555 patients from the German Conn Registry. Dtsch Arztebl Int 2009; 106: 305-311
- 13 Schilbach K, Junnila RK, Bidlingmaier M. Aldosterone to renin ratio as screening tool in primary aldosteronism. Exp Clin Endocrinol Diabetes 2019; 127: 84-92
- 14 Betz MJ, Degenhart C, Fischer E. et al. Adrenal vein sampling using rapid cortisol assays in primary aldosteronism is useful in centers with low success rates. Eur J Endocrinol 2011; 165: 301-306
- 15 Ladurner R, Sommerey S, Buechner S. et al. Accuracy of adrenal imaging and adrenal venous sampling in diagnosing unilateral primary aldosteronism. Eur J Clin Invest 2017; 47: 372-377
- 16 Riester A, Fischer E, Degenhart C. et al. Age below 40 or a recently proposed clinical prediction score cannot bypass adrenal venous sampling in primary aldosteronism. J Clin Endocrinol Metab 2014; 99: E1035-E1039
- 17 Heinrich DA, Adolf C, Holler F. et al. Adrenal insufficiency after unilateral adrenalectomy in primary aldosteronism: Long-term outcome and clinical impact. J Clin Endocrinol Metab 2019; 104: 5658-5664
- 18 Herrada AA, Contreras FJ, Marini NP. et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol 2010; 184: 191-202
- 19 Herrada AA, Campino C, Amador CA. et al. Aldosterone as a modulator of immunity: Implications in the organ damage. J Hypertens 2011; 29: 1684-1692
- 20 Shi Y, Wang H, Su Z. et al. Differentiation imbalance of Th1/Th17 in peripheral blood mononuclear cells might contribute to pathogenesis of Hashimotoʼs thyroiditis. Scand J Immunol 2010; 72: 250-255
- 21 Yoshimoto T, Hirata Y. Aldosterone as a cardiovascular risk hormone. Endocr J 2007; 54: 359-370
- 22 Bendtzen K, Hansen PR, Rieneck K. et al. Spironolactone inhibits production of proinflammatory cytokines, including tumour necrosis factor-alpha and interferon-gamma, and has potential in the treatment of arthritis. Clin Exp Immunol 2003; 134: 151-158
- 23 Munro Neville A. MJOH. Hyperaldosteronism and Related Syndromes of Mineralocorticoid Excess. Springer; London: 1982: 202-241
- 24 Biglieri EG, Schambelan M, Slaton PE. et al. The intercurrent hypertension of primary aldosteronism. Circ Res 1970; 27: 195-202
- 25 Enberg U, Volpe C, Hoog A. et al. Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mRNA expression of the gene CYP11B2. Eur J Endocrinol 2004; 151: 73-85
- 26 Nakamura Y, Felizola SJ, Satoh F. et al. Dissecting the molecular pathways of primary aldosteronism. Pathol Int 2014; 64: 482-489
- 27 Omata K, Satoh F, Morimoto R. et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension 2018; 72: 874-880
- 28 Davis WW, Newsome HH, Wright LD. et al. Bilateral adrenal hyperplasia as a cause of primary aldosteronism with hypertension, hypokalemia and suppressed renin activity. Am J Med 1967; 42: 642-647
- 29 Doorenbos H, Elings HS, Van Buchem FS. Primary aldosteronism due to adrenocortical hyperplasia. Lancet 1956; 271: 335-337
- 30 Ganguly A. Primary aldosteronism. N Engl J Med 1998; 339: 1828-1834
- 31 Katz FH. Primary aldosteronism with suppressed plasma renin activity due to bilateral nodular adrenocortical hyperplasia. Ann Intern Med 1967; 67: 1035-1042
- 32 Ledingham JG, Laragh JH, Sommers SC. Secondary aldosteronism and reduced plasma renin in hypertensive disease. Trans Assoc Am Physicians 1967; 80: 168-182
- 33 Kem DC, Li H, Velarde-Miranda C. et al. Autoimmune mechanisms activating the angiotensin AT1 receptor in ‛primary’ aldosteronism. J Clin Endocrinol Metab 2014; 99: 1790-1797
- 34 Rossitto G, Regolisti G, Rossi E. et al. Elevation of angiotensin-II type-1-receptor autoantibodies titer in primary aldosteronism as a result of aldosterone-producing adenoma. Hypertension 2013; 61: 526-533
- 35 Li H, Yu X, Cicala MV. et al. Prevalence of angiotensin II type 1 receptor (AT1R)-activating autoantibodies in primary aldosteronism. J Am Soc Hypertens 2015; 9: 15-20
- 36 Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 2015; 100: 1-10
- 37 Er LK, Chen L, Tsai YC. et al. Risk of new-onset autoimmune diseases in primary aldosteronism: A nation-wide population-based study. J Hypertens 2019; 38: 745-754
- 38 Piazza M, Seccia TM, Caroccia B. et al. AT1AA (Angiotensin II Type-1 Receptor Autoantibodies): Cause or consequence of human primary aldosteronism?. Hypertension 2019; 74: 793-799
- 39 Turchi F, Ronconi V, di Tizio V. et al. Blood pressure, thyroid-stimulating hormone, and thyroid disease prevalence in primary aldosteronism and essential hypertension. Am J Hypertens 2011; 24: 1274-1279
- 40 Armanini D, Nacamulli D, Scaroni C. et al. High prevalence of thyroid ultrasonographic abnormalities in primary aldosteronism. Endocrine 2003; 22: 155-160
- 41 Cioni K, Pantaleoni M, Toschi E. et al. Exacerbation of autoimmune hypothyroidism after hemi-hypophysectomy in a patient with Cushing's disease. Minerva Endocrinol 1993; 18: 139-141
- 42 Colombo P, Passini E, Re T. et al. Autoimmune thyroid disease after successful treatment of Cushing’s syndrome. J Endocrinol Invest 1994; 17: 289-290
- 43 Takasu N, Komiya I, Nagasawa Y. et al. Exacerbation of autoimmune thyroid dysfunction after unilateral adrenalectomy in patients with Cushing’s syndrome due to an adrenocortical adenoma. N Engl J Med 1990; 322: 1708-1712
- 44 Yamakita N, Sakata S, Hayashi H. et al. Case report: Silent thyroiditis after adrenalectomy in a patient with Cushing’s syndrome. Am J Med Sci 1993; 305: 304-306
- 45 Uthman I, Senecal JL. Onset of rheumatoid arthritis after surgical treatment of Cushing’s disease. J Rheumatol 1995; 22: 1964-1966
- 46 Yakushiji F, Kita M, Hiroi N. et al. Exacerbation of rheumatoid arthritis after removal of adrenal adenoma in Cushing’s syndrome. Endocr J 1995; 42: 219-223
- 47 Noguchi Y, Tamai H, Fujisawa K. et al. Systemic lupus erythematosus after pituitary adenomectomy in a patient with Cushing’s disease. Clin Endocrinol (Oxf) 1998; 48: 670-672
- 48 McGregor AM. Immunoendocrine interactions and autoimmunity. N Engl J Med 1990; 322: 1739-1741
- 49 Murray IP. The effect of prednisolone on Hashimoto’s thyroiditis. Scott Med J 1958; 3: 341-345
- 50 Niepomniszcze H, Pitoia F, Katz SB. et al. Primary thyroid disorders in endogenous Cushing's syndrome. Eur J Endocrinol 2002; 147: 305-311
- 51 Colao A, Pivonello R, Faggiano A. et al. Increased prevalence of thyroid autoimmunity in patients successfully treated for Cushing's disease. Clin Endocrinol (Oxf) 2000; 53: 13-19